(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright McGee, Miller, and Swails 2009
AMBER ADVANCED TUTORIALS
TUTORIAL 3 - SECTION 3.5
MMPBSA.py
Dwight McGee, Bill Miller III, and Jason Swails
0) ノーマルモードの計算のためのセットアップ
2010年5月に示したように、通常モードの計算は、 hereで書かれている通常のインストールプロセス中にnabでコンパイルされるnabプログラム(mmpbsa_py_nabnmode)を介して行われる。このプログラムは実行するために、PATHまたは$AMBERHOME/exeに存在しなければならない。mmpbsa_py_nabnmodeとnmodeの主な違いは、nabプログラムがGeneralized Born solvent中で、通常のモードを計算することができるので、2つ入力変数追加されていることである(マニュアルでnmode_igbとnmode_istrngを参照)。この実装は、以前のバージョンスクリプトで見られる最小化の収束の問題を改善する。1) 出発構造を構築し、平衡化したシステムを得るシミュレーションの実行
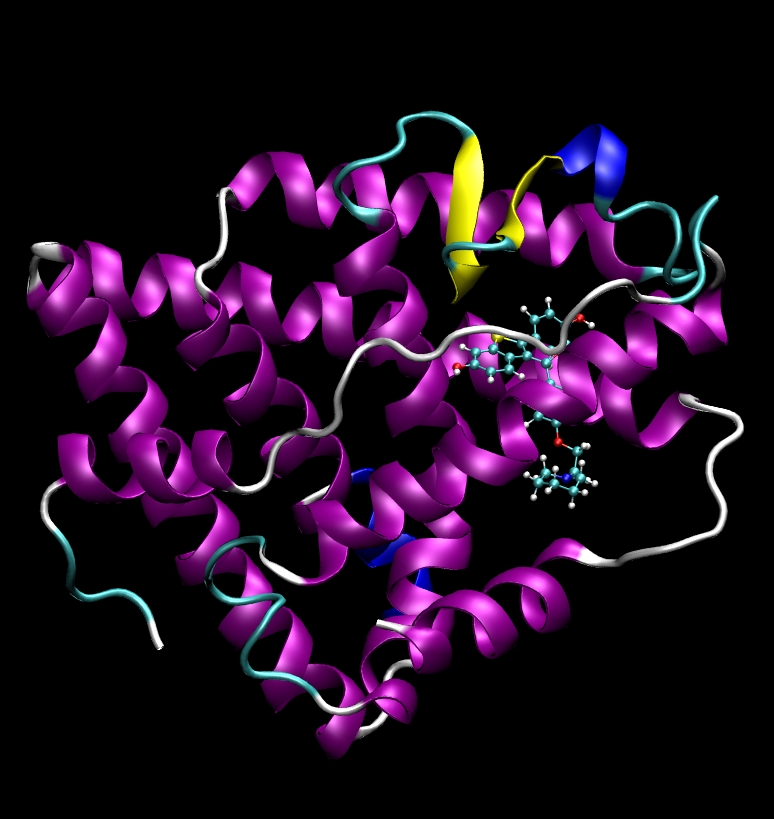
このシミュレーションでモデル化するシステムは、受容体タンパク質 エストロゲンとリガンド ラロキシフェンとの間のタンパク質-リガンド複合体である。下記に、複合体の前準備PDBがある
この構造には、図に示すように、エストロゲン受容体タンパク質にドッキングされたリガンド ラロキシフェンが含まれている。:
このシステムは、Section 1中のRas-Rafシステムと同様に構成されている。出発構造を構築、平衡化システムを得るために、シミュレーションを実行する方法の指示については、Section 1 と Section 2 を参照する。ラロキシフェンのための適切なパラメータを取得するためにantechamberを使用する必要がある。 Sustiva Tutorial (Basic 4) をみる。
MMPBSA.pyを使用して、結合自由エネルギーを計算するためのMD simuationで重要なファイルは、トポロジーファイルとmdcrdファイル(Est_Rec_top_mdcrd.tgz)である。
2) ノーマルモード解析(通常モード)を使用して、結合エントロピーを計算する。
複合体、受容体とリガンドのための通常のモードを計算し、結合エントロピーの推定値を得るために、結果を平均する。システムのエントロピーの寄与はAmberToolsのptrajプログラムを使用してQuasi-Harmonicエントロピー計算を実行するための一般的なセクションの最後の行を非コメントアウトすることによって計算することができることに注意する。
MMPBSA.pyについては、以下の入力ファイルを使用して、通常モードの計算を行う:
| mmpbsa.in |
Input file for running entropy calculations using NMode &general endframe=50, keep_files=2, / &nmode nmstartframe=1, nmendframe=50, nminterval=5, nmode_igb=1, nmode_istrng=0.1, / |
MMPBSA.py用の入力ファイルは、AMBERのsanderモジュールのmdinファイルの設定に同様になるように設計されている。各名前リストは、ampersand (&)で開始し次に名前リストが続く。また、名前リストの終了はbackslash (/)または'&end'で表す。すべての変数の完全なリストは、ユーザーズマニュアルhereを参照する。この入力ファイルは、二つの名前リスト&general と &nmodeに分かれている。入力ファイルは、general, pb, と gbの3つの名前リストに分割されている。&general namelistは、計算の特定の部分の変数を指定するのではなく、すべての部分に変数を指定するように設計されている。このセットアップでは、受容体をRAS、リガンドはラロキシフェンと定義した。'endframe'変数は、ptrajを用いドライmdcrdファイルを作成するときにmdcrdの何フレームで終了するかをセットする。「keep_files'変数は、計算の終了時に削除されるファイルを指定することができる。nmodeセクションは、通常モードの計算に固有の変数を定義するために使用される。'&gb'と '&pb' ネームリストマーカーは、スクリプトでそれらの名前リスト内で定義された指定された値でMM-GBSAとMM-PBSA計算を実行するかを決めている 「nmstartframe '変数は、通常モード解析が始まるフレームをドライmdcrdから定義する。「nmendframe 'と' nmintervalはそれぞれドライmdcrdのノーマルモード解析のためのstartframe、endframeとintervalフレームをセットアップする。nmstartframe/endframe/間隔変数は、startframe、endframe、intervalに従って抽出されたスナップショットの「trajectory」に対応している。したがって、これらのフレームのサブセットだけが(最大でも_MMPBSA_complex.mdcrdの各フレーム)通常モードの計算のために選択される。
$AMBERHOME/bin/MMPBSA.py -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -sp 1err.solvated.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y *.mdcrd > progress.log
または、PBSのように、キューイングシステムにスクリプトを使えば、
qsub nmode.job
スクリプトnmode.jobは、bashシェルでは下記のようになる。 :
| nmode.job |
#!/bin/sh
#PBS -N nmode
#PBS -o nmode.out
#PBS -e nmode.err
#PBS -m abe
#PBS -M email@domain.edu
#PBS -q brute
#PBS -l nodes=1:surg:ppn=3
#PBS -l pmem=1450mb
cd $PBS_O_WORKDIR
mpirun -np 3 MMPBSA.py.MPI -O -i mmpbsa_nm.in -o FINAL_RESULTS_MMPBSA.dat -sp 1err.solvated.prmtop -cp complex.prmtop \
-rp receptor.prmtop -lp ligand.prmtop -y 1err_prod.mdcrd > progress.log
|
これは、progress_nm.logとして計算の進行中に保存される。計算中のすべてのエラーは、同様に、このファイルに出力される。最後に計算完了した後で、スクリプトの各ステップの間に要した時間出力される。シリアルで実行する場合は、この計算を実行すると、10フレームで約40時間かかる。(MMPBSA.py.MPIを並列に実行するできることに注意する。詳細についてはSection 3.4で説明したようにスレッド数が開始時に均等にフレーム数を分割する。システムの規模が大幅に計算時間と必要なメモリ(RAM)に影響を与える。セグメンテーション·フォールト(セグメンテーションフォールト)は、通常、システムに十分なRAMがない場合発生する。
| progress_nm.log |
ptraj found! Using /share/local/lib/amber10/i686/bin/ptraj sander found! Using /share/local/lib/amber10/i686/bin/sander (serial only!) nmode found! Using /share/local/lib/amber10/i686/bin/nmode Preparing trajectories with ptraj... 50 frames were read in and processed by ptraj for use in calculation. Starting sander calls Starting nmode calculations... Timing: Processing Trajectories With Ptraj: 0.240 min. Total Harmonic nmode Calculation Time: 2363.906 min. Output File Writing Time: 0.018 min. Total Time Taken: 2364.165 min. MMPBSA Finished. Thank you for using. Please send any bugs/suggestions/comments to mmpbsa.amber@gmail.com |
コマンドラインで '-y *.mdcrd'は作業ディレクトリ内の '.mdcrd'で終わるすべてのファイルを読み込み、trajectoryとして解析するために使用することをスクリプトに指示する。。
「keep_files」を2にセットすると、ここにすべての出力ファイル保存する。: Nmode_output.tgz.
スクリプトはptrajを使用して、entropy calculations中に分析されたcoordinateである3つの非溶媒和 mdcrdファイル(複合体、受容体、およびリガンド)を作成する。ノーマルモードの計算は各スナップショットのPDBファイルを使用して実行され、10PDBファイルが複合体、受容体、およびリガンドのためのドライmdcrdファイルから作成される。平均PDBファイルは、entropy == 1ならptrajによって行なわれたquasi-harmonic entropy計算にたいするtrajectoryを整列させるための平均構造として作成される。MMPBSA.pyによって作成されたすべてのファイルは、最終的な出力ファイルを除き、接頭辞「_MMPBSA_ 'で始まる。: FINAL_RESULTS_MMPBSA.dat
| FINAL_RESULTS_MMPBSA.dat |
| Run on Thu Feb 11 12:44:26 EST 2010 |Input file: |-------------------------------------------------------------- |Input file for running entropy calculations using NMode |&general | endframe=50, keep_files=2, |/ |&nmode | nmstartframe=1, nmendframe=50, | nminterval=5, |/ |-------------------------------------------------------------- |Solvated complex topology file: 1err.solvated.prmtop |Complex topology file: complex.prmtop |Receptor topology file: receptor.prmtop |Ligand topology file: ligand.prmtop |Initial mdcrd(s): 1err_prod.mdcrd | |Best guess for receptor mask: ":1-240" |Best guess for ligand mask: ":241" |Ligand residue name is "RAL" | |Calculations performed using 50 frames. |Poisson Boltzmann calculations performed using internal PBSA solver in sander. | |All units are reported in kcal/mole. ------------------------------------------------------------------------------- ------------------------------------------------------------------------------- ENTROPY RESULTS (HARMONIC APPROXIMATION) CALCULATED WITH NMODE: Complex: Entropy Term Average Std. Dev. ----------------------------------------------------------- Translational: 16.9389 0.0000 Rotational: 17.3953 0.0038 Vibrational: 2784.7967 2.3982 Total: 2819.1307 2.3964 Receptor: Entropy Term Average Std. Dev. ----------------------------------------------------------- Translational: 16.9233 0.0000 Rotational: 17.3911 0.0045 Vibrational: 2755.5693 3.0352 Total: 2789.8840 3.0342 Ligand: Entropy Term Average Std. Dev. ----------------------------------------------------------- Translational: 13.2972 0.0000 Rotational: 11.4991 0.0496 Vibrational: 33.7549 0.0442 Total: 58.5511 0.0058 DELTA S total= -30.0192 +/- 3.4064 NOTE: All entropy results have units kcal/mol. (Temperature has already been multiplied in as 300. K) ------------------------------------------------------------------------------- ------------------------------------------------------------------------------- |
出力ファイルの先頭には、計算に関するさまざまな詳述が含まれている。出力ファイルの残りの部分には、転移、回転、振動、及び総エントロピーの寄与の平均値、標準偏差、および平均値の標準誤差、を含む。これらのセクションの後、ΔSは、標準偏差と標準誤差と一緒に記述されている。
一般的には、生物学的複合体には負のΔS値を期待する。タンパク質とリガンドが複合体を作るために結合し、従って可能な微視的状態の減少を意味する。利用可能な微視的状態の減少は主に、リガンドがトラップされタンパク質に結合すると移動度が制限され生じる。
負のΔS値-30.02キロカロリー/モルから、純水中ではエントロピー的に不利なタンパク質-リガンド複合体であるが、複合体形成に有利なenthalpic寄与を推定しなかったので、結果は実際の結合自由エネルギーに等しくないことに注意する。
CLICK HERE TO GO TO SECTION 3.6
CLICK HERE TO GO TO MMPBSA.py HOME
CLICK HERE TO GO BACK TO THE INTRODUCTION
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright McGee, Miller, and Swails 2009