(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2006
AMBER ADVANCED TUTORIALS
TUTORIAL 3 - SECTION 1
MM-PBSA
By Ross Walker & Thomas Steinbrecher
1) 初期構造を構築し、平衡化したシステムを取得するために、シミュレーションを実行する。
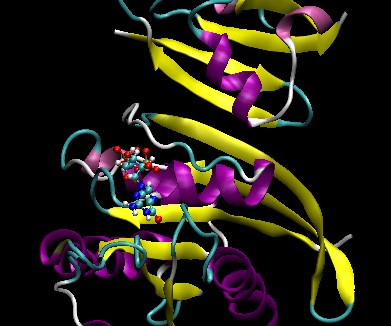
このシミュレーションでモデル化するシステムは、ヒトH-Rasタンパク質とシグナル伝達カスケードの中心であるC-Raf1(RAS-RAF)のラス結合ドメインとの複合体である。ここでは、事前に準備し部分的に平衡化したRAS-RAF複合体のPDBファイルを用いる。
この構造には、以下の図に示すようにrasおよびRAFタンパク質と生理的に必要なGTPヌクレオチドが含まれている。:
このタンパク質中には基本的にGTP分子に結合しているマグネシウムイオンもあるが、削除する。したがって、PDBファイルからの残基243および244を削除する必要がある。
次のステップでは、このPDBファイルを二つの別々の構造ras.pdbとraf.pdbに分割し、都合3個のファイルRAS-raf.pdb、ras.pdbとraf.pdbを得る。次いで、3つのガス相prmtopとinpcrdファイルのペアをMM-PBSA計算に、MDシミュレーションを実行するために使用される溶媒和複合体のための1つのprmtopとinpcrdファイルペアを作成する。 :
Caution: AMBER 14ででFF99力場をtleapにロードするには-f $AMBERHOME/dat/leap/cmd/oldff/leaprc.ff99を使用する。> $AMBERHOME/bin/tleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff99
計算方法の正しい半径を選択していることを確認する。詳細については、マニュアルのLEaP sectionのPBRadii項セットを参照する here。.com = loadpdb ras-raf.pdb
ras = loadpdb ras.pdb
raf = loadpdb raf.pdb
set default PBRadii mbondi2
saveamberparm com ras-raf.prmtop ras-raf.inpcrd
saveamberparm ras ras.prmtop ras.inpcrd
saveamberparm raf raf.prmtop raf.inpcrd
tleapを終了する前に、MDシミュレーションを実行するための溶媒和複合体を作成する必要がある。:
charge com
> Total unperturbed charge: -0.000000
> Total perturbed charge: -0.000000 (Hence there is no need to add counter ions)
solvatebox com TIP3PBOX 12.0
saveamberparm com ras-raf_solvated.prmtop ras-raf_solvated.inpcrd
quit
必要なファイルは以下からもダウンロードできる。: ras-raf.prmtop, ras-raf.inpcrd, ras.prmtop, ras.inpcrd, raf.prmtop, raf.inpcrd, ras-raf_solvated.prmtop, ras-raf_solvated.inpcrd
1.1) 溶媒和複合体の平衡化
短い最小化、弱い拘束で50ピコ秒の加熱および50ピコ秒密度の平衡の後、300Kで一定の圧力平衡化を500PSを行うことにより、溶媒和複合体を平衡化する。すべてのシミュレーションは、水素原子でシェイクして、温度制御のための2fs時間ステップとランジュバン力学を実行する。入力ファイルは次のとおりである。:
| minimise ras-raf &cntrl imin=1,maxcyc=1000,ncyc=500, cut=8.0,ntb=1, ntc=2,ntf=2, ntpr=100, ntr=1, restraintmask=':1-242', restraint_wt=2.0 / |
heat ras-raf &cntrl imin=0,irest=0,ntx=1, nstlim=25000,dt=0.002, ntc=2,ntf=2, cut=8.0, ntb=1, ntpr=500, ntwx=500, ntt=3, gamma_ln=2.0, tempi=0.0, temp0=300.0, ntr=1, restraintmask=':1-242', restraint_wt=2.0, nmropt=1 / &wt TYPE='TEMP0', istep1=0, istep2=25000, value1=0.1, value2=300.0, / &wt TYPE='END' / |
| heat ras-raf &cntrl imin=0,irest=1,ntx=5, nstlim=25000,dt=0.002, ntc=2,ntf=2, cut=8.0, ntb=2, ntp=1, taup=1.0, ntpr=500, ntwx=500, ntt=3, gamma_ln=2.0, temp0=300.0, ntr=1, restraintmask=':1-242', restraint_wt=2.0, / |
heat ras-raf &cntrl imin=0,irest=1,ntx=5, nstlim=250000,dt=0.002, ntc=2,ntf=2, cut=8.0, ntb=2, ntp=1, taup=2.0, ntpr=1000, ntwx=1000, ntt=3, gamma_ln=2.0, temp0=300.0, / |
| Caution: このチュートリアルの例では、乱数発生器に使用されるランダムシード値を変更しない。これは、変数ネームリストigによって制御されており、チュートリアルの設定範囲での結果の再現性を得るためである。最終的に長い時間のシミュレーションを実行している場合、特にNTT = 2または3(アンダーソンかランジュバンサーモスタット)を使用する場合は、MD再起動時に常にデフォルト値から乱数シードを変更しなければならない。AMBER 10(bugfix.26以降)またはAMBER 11以降を使用している場合は、ig=-1設定することで、その変更は自動的に行うことができる。それ以外では、計算を再起動するたびにigに正の乱数を指定しなければならない。この作業をしない場合生じる問題の詳細は、以下のマニュアルを参照する。: |
コマンドを使用して、これらの4つのシミュレーションのすべてのを実行する必要がある。” \”で区切られているのは続きの行であることに注意する:
$AMBERHOME/bin/sander -O -i min.in -o min.out -p ras-raf_solvated.prmtop -c ras-raf_solvated.inpcrd \
-r min.rst -ref ras-raf_solvated.inpcrd
$AMBERHOME/bin/sander -O -i heat.in -o heat.out -p ras-raf_solvated.prmtop -c min.rst \
-r heat.rst -x heat.mdcrd -ref min.rst
gzip -9 heat.mdcrd
$AMBERHOME/bin/sander -O -i density.in -o density.out -p ras-raf_solvated.prmtop -c heat.rst \
-r density.rst -x density.mdcrd -ref heat.rst
gzip -9 density.mdcrd
$AMBERHOME/bin/sander -O -i equil.in -o equil.out -p ras-raf_solvated.prmtop -c density.rst \
-r equil.rst -x equil.mdcrd
gzip -9 equil.mdcrd
これは、1.7GHz、IBM P690の16プロセッサで約5時間かかる。
ここでは、出力ファイルは、 equil.tar.gz。
MM-PBSAproductionMDの実行を続ける前にシステムが平衡化したことを確認する必要があるので、温度、密度、全エネルギーとRMSDを検討する。次のPerlスクリプト(process_mdout.pl)を使用し出力ファイルから有用な情報を抽出する。
./process_mdout.pl heat.out density.out equil.out
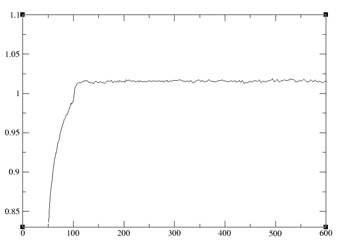
最初の加熱を一定容量の条件下で行ったので、濃度データが記録されていない。従ってsummary.DENSITYファイルを編集して、最初の50行を削除する必要がある。(xmgraceは、50行の削除を行わないとグラフをまともに描けない)。
xmgrace summary.DENSITY
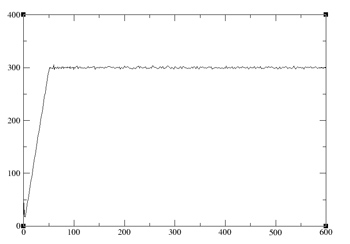
xmgrace summary.TEMP
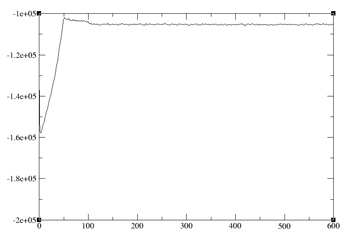
xmgrace summary.ETOT
さらに、立体配座の安定性が平衡になっているかを確認するためにエネルギー最小化構造時のタンパク質骨格のRMSDを検討する。これは、次のスクリプトでptrajまたはcpptrajを使用して行う。:
| measure_equil_rmsd.ptraj |
| trajin equil.mdcrd.gz 1 250 1 reference ras-raf_solvated.inpcrd rms reference out equil.rmsd @CA,C,N |
xmgrace equil.rmsd
 DENSITY |
 TEMPERATURE |
 TOTAL ENERGY |
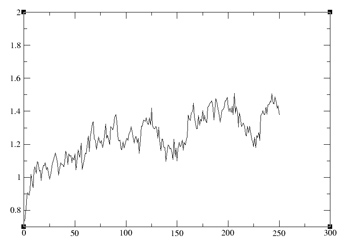 BACKBONE RMSD |
密度、温度および総エネルギーの時間に対するプロットは、それらが平衡化していることを示した。RMSDは、一定のレベルに到達するかに見えるが、完全に収束したとは言えない。しかしこのチュートリアルの目的のためには許容範囲にある。実際の計算では、システムに応じて、より長い平衡時間をかけたい。次にproduction runsを実行する。
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2006