Tutorial A24
Free energy calculations with the Free Energy Workflow Tool (FEW)
By: Nadine Homeyer and Holger Gohlke
Please note: ここで紹介するワークフローツールのいくつかはAmberTools 14またはそれ以降のバージョンで利用可能で、AmberToolsとAMBERバージョン14以降で動作する。AmberToolsとAMBER 14は、ガイドライン AMBER manualに従ってインストールすることができる。バグフィックスと新しいソースコードがインストールプロセスの過程で自動的にダウンロードされる。
このチュートリアルでは、同じ受容体に結合するリガンドセットの自由エネルギー計算を自動セットアップするワークフローツールFEWの機能を示す。FEWは分子力学ポアソン-ボルツマン表面積(MM-PBSA)に応じた自由エネルギー計算、分子力学generalized Born表面積(MM-GBSA)、線形相互作用エネルギー(LIE)、および熱力学的相互作用(TI )アプローチの準備する。FEW使用する前に、AMBERを使用する基本的な分子動力学(MD)シミュレーションの実行、適用したい自由エネルギー計算のタイプの理論とセットアップを理解している必要がある。MM-PB(GB)SAとTIのアプローチにより、分子動力学シミュレーションと自由エネルギー計算の設定と実行を示すチュートリアルは
自由エネルギー計算手法の理論の説明は、次のレビューや書籍にある:MM-PB(GB)SA [1,
2],
LIE [3,
4],
and TI [5,
6]。さらに、次の記事も参考になる N.
Homeyer, H. Gohlke, FEW - A Workflow Tool for Free Energy Calculations of Ligand
Binding. J. Comput. Chem.
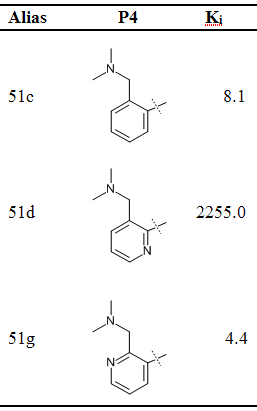
2013, 34, 965–973。このチュートリアルに示す自由エネルギー計算サンプルは、その資料に記載事例研究から選択した。血液凝固カスケードにおいて重要な機能を果たすプロテアーゼであるタンパク質Xa因子を阻害する3リガンド(表1)の相対的結合自由エネルギーを決定する。
訳者註:amber14のマニュアル29 MMPBSA.pyに以下のような説明がある。
29. MMPBSA.py
29.2. MM/PB(GB)SA 計算の準備
リガンドを比較しその相対的結合自由エネルギーを得るためにMM/PB(GB)SA は大変便利なツールである。その最大の特徴は、 TI (Thermodynamic Integration) や FEP (Free Energy Perturbation) に比べて コンピューターに負担がかからないことである。MD simulationを行う前に下記のアドバイスに従えば、簡単に MMPBSA.py を実行し良い結果を得られる。
29.2.1.Topology Files の作成
MMPBSA.pyは少なくとも3つ、普通は4つのコンパチブルなtopologyファイルを必要とする。 もしMDを露わな水(explicit water)中で行うなら、溶媒和したtopology fileを複合体に用意し、さらに複合体、受容体、リガンドそれぞれにtopologyファイルを用意する必要がある。さらにそのファイルは相互にコンパチブルでなければいけない(それぞれは、同じ原子に関して同じ荷電をもち、それぞれのprmtopに対して同じforce field を使用し、同じPBRadii setを持っている必要がある)。これに関してはLeapのpbradiiの説明を参照。
「溶媒効果を扱う方法には, 溶媒分子をあらわに取り扱う explicit solvent 条件 と,平均的な溶媒環境をパラメータとして近似的に取り扱う implicit solvent 条件がある (東北薬科大学研究誌,57,67−72(2010))」
必要な入力構造:
| Table 1:Ligand structures and experimentally determined Kivalues in [nM] for Factor Xa |
| |
準備
このチュートリアルで容易にFEWを使うために、パス変数$FEWにシステムのFEWがインストールされているフォルダーを設定する。例えば、/opt/amber14/Ambertools/src/FEWにインストールされている場合、:1. 分子動力学(MD)シミュレーションのセットアップ
MM-PB(GB)SAとLIE計算はMDシミュレーションから得られるスナップショットに基づいたFEWで実行される。従ってこのステップは これらのタイプの自由エネルギー計算に先立って実行される必要がある。TI計算は結晶構造から直接開始できるが、不適切なコンタクトを取り除くために まず短いMD平衡化を行うことを推奨する。 ここでは、後者の選択肢に沿って実行する。 前者のオプションについては、AMBER manualのFEWセクション参照する。MDシミュレーションは、ここでは2つのステップでセットアップする。最初はリガンドのためのパラメータを決定し(ステップ1A)、次にMDシミュレーション用の入力ファイルを生成する(ステップ1B)。必要な入力情報及びFEWによって生成された出力を伴う2つのステップは、図1に示されている。
注意: デモ目的のシミュレーションのためのこのチュートリアルでは、複合体、受容体、およびリガンド(3-軌道・座標アプローチ)の設定となる。1軌道・座標アプローチ(すなわちLIEまたはTIの計算が無い)のみに従ってMM-PB(GB)SAの計算を行う場合は、この時点で複合体シミュレーションのセットアップだけすることを勧める。 A:
パラメータ・ファイルの作成
注:: このチュートリアルでは、最も簡単な手続き;つまりAM1-BCCの手順で決定された原子電荷を用いてシミュレーションの準備をすることが示した。RESP荷電を用いてシミュレーションをセットアップをしたい場合、マニュアルを参照し、$FEW/examples/command_files/commonMDsetup/leap_resp_step1
と$FEW/examples/command_files/commonMDsetup/leap_resp_step2を使用しなさい。
B:
MDの入力ファイルの作成 MDシミュレーションのセットアップ:: 平衡化: 平衡化はequiディレクトリにある
テンプレートファイルを用いて準備する。ここで使用される平衡化手順において、分子システムは、まず、第1の高い拘束 (min_ntr_h.in)
、次に低い拘束(min_ntr_l.in)
が受容体および/またはリガンド、すなわち、溶質に適用される。受容体の場合には1からの全ての残基(no_of_rec_residues )を拘束する。最小化後、MD equilibrationsは、合計400psに渡って溶質を低拘束にて行う。まず温度がNVT条件下300 Kまで上昇する(md_nvt_ntr.in) 、次いで密度が1g/cm3に調整されている NPTシミュレーション(md_npt_ntr.in )、 そして最終的にNVTシミュレーション (md_nvt_red_<number>.in).で溶質に対する制約を50 psで除去する。 Production: MDのProductionのための入力ファイルをテンプレートファイル
MD_prod.inに基づいて作成し総時間が2ナノ秒まで達するようにする。この短いシミュレーション時間は、デモンストレーションのみを目的として選択した。普通はるかに長いシミュレーション時間が、代表的スナップショットを抽出することが可能、完全に平衡化した状態の軌道を得るために必要とされる。
平衡のファイルrestart_file_for_MDprod からの最終座標ファイルはmd_nvt_red_06でありMD production inputに使われる。 MDの生産のためのテンプレートバッチファイル MDprod_batch_templateを固定変数ステートメントとRe-queueセクションのMDシミュレーションを実行するコンピューティングアーキテクチャより前もって調整する必要がある。
perl $FEW/FEW.pl MMPBSA /home/user/tutorial/cfiles/setup_am1_3trj_MDs MDの平衡化を開始するには、MD_am1フォルダのqsub_equi.shシェルスクリプトを実行する。
全てのリガンド/複合体で平衡化が完了すると、/font>
MD_am1フォルダでそれぞれのシェルスクリプトを呼び出すことでproductionを開始する。

図 1:FEW とMDシミュレーションのセットアップの概略図。
このチュートリアルで使用する入力情報を青で強調表示、他のシステム/パラメータの設定のために必要な入力データは、灰色で表示、コマンド・ファイルは、赤でマークした。ここで使用してない入力の例(上記スキームの灰色)は、$FEW/examples/input_infoフォルダにある。必要なデータフォーマットの説明はAMBERマニュアルのFEWセクションで提供されている。
leap_am1
@WAMM
################################################################################
# Location and features of input and output directories / file(s)
#
# lig_struct_path: Folder containing the ligand input file(s)
# multi_structure_lig_file: Basename of ligand file, if multi-structure
# file
is provided
# output_path: Basis directory in which all setup and analysis
# folders
will be generated
# rec_structure: Receptor structure in PDB format
# bound_rec_structure: Optional, alternative receptor structure in bound
# conformation
to be used for 3-trajectory approach
lig_struct_path
./tutorial/structs
multi_structure_lig_file
output_path
./tutorial
rec_structure
./tutorial/input_info/2RA0_IN.pdb
bound_rec_structure
# Specification of ligand input format
lig_format_sdf 0
lig_format_mol2 1
# Receptor features
# water_in_rec: Water present in receptor PDB structure
water_in_rec 1
# Request structure separation
# structure_separation: Separate ligands specified in one
multi-structure
#
input file and generate one structure file per ligand.
structure_separation 0
################################################################################
# Creation of LEaP input files
#
# prepare_leap_input: Generate files for LEaP input
# non_neutral_ligands: Total charge of at least one molecule is not zero.
#
In this case the total charge
of each non-neutral
#
molecule must be defined in
lig_charge_file.
# lig_charge_file: File with information about total charges of ligands.
# am1_lig_charges: Calculate AM1-BCC charges.
# resp_lig_charges: Calculate RESP charges. In this case charges must be
# computed
with the program Gaussian in an intermediate step.
# Batch
scripts for Gaussian calculations will be prepared
# automatically,
if requested (see prepare_gauss_batch_file
# and
gauss_batch_template below).
# calc_charges: Calculate charges according to requested procedure.
# If
this flag is set to zero, no atomic charges are calculated.
# resp_setup_step1: Step 1 of RESP charge calculation: Preparation of
# Gaussian
input.
# resp_setup_step2: Step 2 of RESP charge calculation: Generation of
LEaP input
# from
Gaussian output.
# prepare_gauss_batch_file: Generate batch script for Gaussian input if
RESP
#
charge calculation is
performed. A batch template
#
file
(gauss_batch_template) is
required.
# gauss_batch_template: Batch template file
#
Prerequisite: resp_lig_charges=1, resp_setup_step1=1
#
and prepare_gauss_batch_file=1
# gauss_batch_path: Basic working directory for Gaussian jobs
# average_charges: If the charges of two steroisomers shall be averaged, so that
#
both ligands obtain the same atomic charges, a file in which
#
the stereoisomer pairs are specified must be given here.
prepare_leap_input 1
non_neutral_ligands 0
lig_charge_file
am1_lig_charges 1
resp_lig_charges 0
calc_charges 1
resp_setup_step1 0
resp_setup_step2 0
prepare_gauss_batch_file 0
gauss_batch_template
gauss_batch_path
average_charges
leap_am1
の ファイルを直接FEWの入力コマンド・ファイルとして使用することができる。赤で示した
基本的な input/output path
は使用しているシステム上のチュートリアルフォルダのpathに応じて変更する。
分かりやすいようにコマンドファイル中の comments
は緑字で示し、 key
terms とparameters は黒字で示してある。
ファイルの最初の部分では、一般的な入出力データを定義するために必要なパラメータを指定した。このチュートリアルでは、単一のMOL2構造を使用するので、
、multi_structure_ligand_file を設定する必要はない。bound_rec_structureは 、このセットアップでは結合複合体および遊離受容体に単一のタンパク質構造を使用するので指定していない。リガンド結合の際に、受容体のコンフォメーションが変わる場合は、二つの異なる受容体構造、結合状態bound_rec_structure
と自由状態rec_structure, を使える。ここで使用する受容体構造体は、Protein
Data Bank で 2RA0として引用できる。この構造体に存在する分離結晶水分子は受容体モデル構造2RA0_IN.pdbにHOH残基エントリとして指定されているので、water_in_rec フラグが1に設定されている。
コマンドファイルの2番目の部分は、LEAPのための基本的な入力ファイルの生成に必要なパラメータを含む 。ここでAM1-BCCprocedure によるリガンドの原子電荷計算が実行され、従ってRESPprocedureによる原子電荷の計算に必要とされるガウスの計算に関連する全てのパラメータは必要ない。さらに全てのリガンドは全体として中性であり(non_neutral_ligands=0)、全体のリガンドの電荷を決めるどのlig_charge_fileも指定する必要はない 。
FEW実行:
tutorial
folderの存在場所に応じてleap_am1ファイルの赤でマークされたパスを変更した後、 荷電計算、パラメータの調製ステップを起動する。
:
perl
$FEW/FEW.pl MMPBSA ./tutorial/cfiles/leap_am1
原子電荷の計算は計算負荷が高いため、このコマンドの実行はワークステーションで約半時間かかる。
出力:
この最初の手順が正常に終了した場合には、新しいleapフォルダが./tutorialに作られる。leap
フォルダには各リガンドのサブフォルダがあり、Leapを用いるMDシミュレーションのセットアップに必要な構造とパラメータファイルを含む。
setup_am1_3trj_MDs
@WAMM
################################################################################
# Location and features of input and output directories / file(s)
#
# lig_struct_path: Folder containing the ligand input file(s)
# multi_structure_lig_file: Basename of ligand file, if multi-structure
#
file
is provided
# output_path: Basis directory in which all setup and analysis
# folders
will be generated
# rec_structure: Receptor structure in PDB format
# bound_rec_structure: Optional, alternative receptor structure in bound
#
conformation to be used for
3-trajectory approach
lig_struct_path
/home/user/tutorial/structs
multi_structure_lig_file
output_path
/home/user/tutorial
rec_structure
/home/user/tutorial/input_info/2RA0_IN.pdb
bound_rec_structure
# Specification of ligand input format
lig_format_sdf 0
lig_format_mol2 1
# Receptor features
# water_in_rec: Water present in receptor PDB structure
water_in_rec 1
################################################################################
# Setup of molecular dynamics simulations
#
# setup_MDsimulations: Perform setup of simulation input
# traj_setup_method: 1 = One trajectory approach
#
3 = Three trajectory approach
# MD_am1: Prepare simulations with AM1-BCC charges
# MD_resp: Prepare simulations with RESP charges
# SSbond_file: File with disulfide bridge definitions
# additional_library: If an additional library file is required, e.g. for
#
non-standard residues present
in the receptor structure,
#
this file must be specified
here.
# additional_frcmod: If additional parameters are needed, e.g. for describing
#
non-standard residues present in the
receptor structure,
#
a parameter file should be provided here.
# MD_batch_path: Path to basis directory in which the simulations shall
#
be performed in case this differs from <output_path>.
#
If no path is given, it is assumed that the path is
#
equal to <output_path>
# MDequil_template_folder: Path to directory with equilibration template files
# total_MDequil_time: Total equilibration time in ps
# MDequil_batch_template: Batch template file for equilibration
# MDprod_template: Template file for production phase of MD simulation
# total_MDprod_time: Number of ns to simulate
# MDprod_batch_template: Batch template file for MD production
# no_of_rec_residues: Number of residues in receptor structure
# restart_file_for_MDprod: Base name of restart-file from equilibration that
#
shall
be used for production input
setup_MDsimulations 1
traj_setup_method 3
MD_am1 1
MD_resp 0
SSbond_file /home/user/tutorial/input_info/disulfide_bridges.txt
additional_library
/home/user/tutorial/input_info/CA.lib
additional_frcmod
MD_batch_path
MDequil_template_folder /home/user/tutorial/input_info/equi
total_MDequil_time 400
MDequil_batch_template /home/user/tutorial/input_info/equi.pbs
MDprod_template /home/user/tutorial/input_info/MD_prod.in
total_MDprod_time 2
MDprod_batch_template /home/user/tutorial/input_info/prod.pbs
no_of_rec_residues 290
restart_file_for_MDprod md_nvt_red_06
入力/出力情報
Input/Output
information:
入力と出力データのパラメータが指定されているコマンドファイルの一部は、step 1A準備に示したものと同等である。この部分は要求された機能とは関係なく、すべてのコマンドファイルで定義する必要がある。さもないと FEWは入力ファイルをどこに配置しどう処理するか、出力を書き込む場所を決められない。
シミュレーションは、AM1-BCC電荷を使用して、3-軌道法に従って実行する。第Xa因子タンパク質に存在するジスルフィド架橋は、SSbond_fileのジスルフィド結合のペア残基をタブで分けたリストの形でdisulfide_bridges.txtで指定される。ライブラリファイル additional_library
は、 受容体構造内に存在する構造的に結合したカルシウムイオンのために必要である。カルシウムのためのパラメータは、受容体構造の記述のためにデフォルトで使用されるff12SB力場で利用できるので、追加のパラメータは必要ない。
MD の入力ファイルが処理される順序は、下の指定したバッチスクリプトMDequil_batch_templateで定義される。
別の平衡化手順を使用する場合は、平衡テンプレートファイルを修正する必要があり、sanderは バッチファイルで呼び出す。しかし、ここで適用される通常の手順は、完全に平衡化した分子系をもたらすはずである。(この平衡化手順のテンプレートファイルもFEWチュートリアルと共に$FEW/examples/input_info/equiで提供されている。) MDequil_batch_template におけるコンピューティング・アーキテクチャ固有の設定スクリプトは、一般的に、ローカルコンピューティング施設の要件に応じて適合させる必要がある。上記の平衡化手順を使用したい場合は、スクリプトのFix variablesまでの最初の部分だけを変更する。
今更 NVTとNPT:高分子系のシミュレーションで有用なアンサンブルは、実験測定との比較に便利なNVTアンサンブル(N(粒子数)、V(体積)、T(温度)を一定に保つ系)やNPTアンサンブル(N、P(圧力)、Tを一定に保つ系)である。NVTアンサンブルのシミュレーションでは熱浴と接触して温度を一定に保つ分子動力学計算、NPTアンサンブルのシミュレーションでは圧力溜と接触して圧力を一定に保つ分子動力学計算が行われ、種々の熱力学量が求められる。(構造生物 Vol.5 No.3 広野、合田)
FEW実行:
基本的な入力/出力パスbasic input/output pathが 正しく指定されたことを確認した後 、MDシミュレーションのセットアップはFEWを呼び出すことができる。:
出力:
FEW 実行が正常に完了した場合には、
basic input/output directoryすなわち、 チュートリアル フォルダに新しいフォルダ MD_am1(図2)が作られる。MDフォルダの名前が示しているように、AM1-BCCの電荷を使って分子動力学シミュレーションを実行するためのファイルが含まれている。MD_am1フォルダには、各リガンドと受容体に対するサブフォルダ(rec)がある。 すべてのサブフォルダは、初期座標とトポロジーファイルが存在するcrystフォルダとcom(for complex) とlig(for ligand)フォルダが存在する。 後者のフォルダは、それぞれのシステムに対するequi(with all files for MD equilibration)とprod(with the files for MD production) を含んでいる。
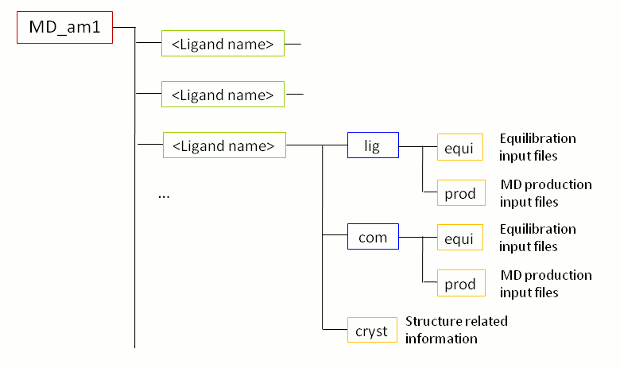
図2:
ステップ1(b)にFEWによって作成されたMDフォルダーの下部構造の模式図。
手元のMDシミュレーションから軌道で、それは自由エネルギー計算のセットアップを継続することができる。
興味があるセクションに移動するには、以下のリンクのいずれかをクリックする。
SECTION 2: MM-PB(GB)SA calculations
Copyright: Nadine Homeyer 2014