![]()
Tutorial B4
Using Antechamber to Create Leap Input Files for Simulating
Sustiva (efavirenz)-RT complex
using the General Amber Force Field
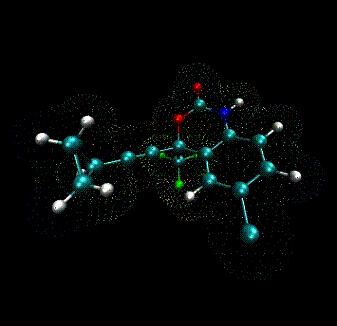
By Ross Walker and Sishi Tang
訳
大阪府立大学・生命環境科学研究科
応用生命科学専攻・生体情報化学研究室
和田野 晃
Introduction
Antechamber は"general AMBER force field (GAFF)1)"とともに使用する。 このforce field は特にほとんどのpharmaceutical molecules をカバーする様に作成され、 traditional AMBER force fields と互換性があり、シミュレーションでは一緒に用いること が可能である。traditional AMBER force fieldsと同様に, GAFFは結合と結合角にたいして a simple harmonic function formを用いるが、 一般のタンパク質 と DNA に対応した AMBER force fields とは異なりGAFF で使用されている原子種はより一般的で、ほとんどの有機化合物に 含まれる原子をカバーしている。現在、GAFF force field は33 basic atom types と 22 special atom typesを含んでいる。 charge methods としては HF/6-31G* RESP2) or AM1-BCC3)が使用可能である。
目的的に, GAFFは完全なforce field で、パラメータが存在しない場合はまれであり、有機化学で現れる C, N, O, S, P, H, F, Cl, Br および Iをカバーしている。さらに、GAFF は、AMBER macromolecular force fieldsと完全に互換性があり、合理的ドラッグデザインに対する分子動力学的 ツールになるであろう。特に、結合自由エネルギーの計算や分子ドッキング研究では重要になると思われる。
Antechamber tool set は、AMBER simulation programs に使用するtopology files を短時間で作れるように設計されている。 以前のtutorialでは、マニュアルで原子タイプをアサインしていたが、 antechamber はGAFF により自動的にアサインすることを可能に している。Antechamberは以下の問題を解決する。:
Automatically identify bond and atom types
Judge atomic equivalence
Generate residue topology files
Find missing force field parameters and supply reasonable suggestions
しかし、Antechamber は必ずしも完全に上記の問題を解決するわけではない。従って、常にAntechamberがアサインした原子タイプを自分 で注意深くチェックする必要がある。Scientific softwareを内容がわからないまま、"Black Box" として使用しないようにする。
------------------------------------------------------------------------
TUTORIAL B4 - SECTION 2
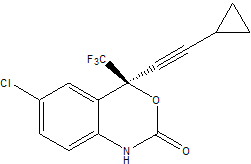
早速、antechamber を sustiva pdb fileに適応してみる。leapで新しいunitを定義するために必要な"mol2" file を作成するのにまず下記のcommandを使う。:
antechamber -i sustiva_new.pdb -fi pdb -o sustiva.mol2 -fo mol2 -c bcc -s 2
Running: /usr/local/amber10/bin/bondtype -j full -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac |
最後には次のファイルが作業ディレクトリに作られる。
ANTECHAMBER_AC.AC ANTECHAMBER_AM1BCC_PRE.AC ATOMTYPE.INF mopac.out sustiva.mol2 |
大文字で示すファイルはantechamberが使う中間ファイルであり、以降では必要がないので削除してもよい。うまくいかない場合、チェックすることが出来るので初期設定では削除しない。mopac.xxx files はAntechamber がatomic point charges を計算するために用いるmopac quantum mechanics code のinputとoutputファイルである。ここでは、mopac calculationが完全に実行されたかどうかをチェックする以外には用いない。:
*******************************************************************************
** FRANK J. SEILER RES. LAB., U.S. AIR FORCE ACADEMY, COLO. SPGS., CO. 80840 ** ******************************************************************************* AM1 CALCULATION RESULTS ******************************************************************************* * MOPAC: VERSION 6.00 CALC'D. * GEO-OK - OVERRIDE INTERATOMIC DISTANCE CHECK * MMOK - APPLY MM CORRECTION TO CONH BARRIER * ANALYT - USE ANALYTIC DERIVATIVES * * * * CHARGE ON SYSTEM = 0 * * * * T= - A TIME OF 3600.0 SECONDS REQUESTED * DUMP=N - RESTART FILE WRITTEN EVERY 3600.0 SECONDS * AM1 - THE AM1 HAMILTONIAN TO BE USED * PRECISE - CRITERIA TO BE INCREASED BY 100 TIMES ***********************************************************************100BY100 AM1 ANALYT MMOK GEO-OK PRECISE CHARGE=0 created by wmopcrt() for mopac |
ここで興味あるファイルは、susutiva.mol2で、Antechamberの実行はこのファイルを作成するためである。(sustiva.mol2 file) このファイルはsustiva残基の定義を含んでおり、prmtopとinpcrdファイルを作成するときにチャージやatom typeの情報を今ファイルからleapに読み込む。このファイルをさっと見ておくと参考になる。:
@<TRIPOS>MOLECULE SUS 30 32 1 0 0 SMALL bcc @<TRIPOS>ATOM 1 CL -4.6850 -32.7250 25.2220 cl 999 SUS -0.073100 2 F1 -0.7550 -36.6320 25.6970 f 999 SUS -0.231400 3 F2 1.0780 -37.0430 24.6720 f 999 SUS -0.221400 4 F3 -0.7840 -37.1770 23.6260 f 999 SUS -0.216800 5 O1 1.5240 -34.9340 20.9100 o 999 SUS -0.573600 6 O2 0.9890 -34.8800 23.0580 os 999 SUS -0.371900 7 N -0.6810 -34.9710 21.4340 n 999 SUS -0.459500 8 C1 -1.6620 -34.4140 22.3130 ca 999 SUS 0.084700 9 C2 -2.9150 -33.9470 21.8430 ca 999 SUS -0.167000 10 C3 -3.8380 -33.4230 22.7710 ca 999 SUS -0.069500 11 C4 -3.5330 -33.3730 24.1190 ca 999 SUS -0.025500 12 C5 -2.3100 -33.8290 24.5930 ca 999 SUS -0.040200 |
このファイルには、sustiva残基の全てのチャージ、原子タイプ、が含まれ、それらにより、Leap を使ってprmtopとinpcrdファイルを作ることになる。 このファイルには、sustiva 分子の3D構造、それぞれの原子上のチャージ、原子の番号(column 1)、名前 (column 2) アトムのタイプ (column 6)が含まれている。 しかし、このファイルにはパラメータは含まれていない。GAFF parameters は全て$AMBERHOME/dat/leap/parm/gaff.datに書かれている.。またここで示されているGAFF atom typesは、全て小文字で表されている。これは、GAFF force field がmacromolecular AMBER force fieldsと独立して扱われるためである。普通、 AMBER force fields は大文字の原子タイプで示され、GAFF と traditional force fields が同じ計算のなかで混同されないようになっている。
ほとんどのbond, angle と dihedral parametersパラメータは、このファイル中で定義されてはいるが、まだ定義されずに残っているものがある可能性もある。その場合、定義されていないパラメータをprmtopとinpcrdファイルをLeapで作成する前に調べておく必要がある。そのために、parmchkユーティリティを用いる。
parmchk -i sustiva.mol2 -f mol2 -o sustiva.frcmod
このコマンドを実行し、sastiva.frcmodファイルを作成する。このパラメータファイルは、 Leapを実行する際に定義されてないパラメータを加えるために使用する。antechamber は、missing parametersを 同様のパラメータに対する analogy で可能な限り作成する。しかし、シミュレーションの前に、これらのパラメータを少し注意深くチェックする必要がある。Antechamber が経験的にそれらの値を計算することが不可能であったり、アナロジーが無理な場合は、最もリーゾナブルだと思われる初期値を記入するか、ゼロを入れ"ATTN: needs revision"というコメントを書き加えるからである。その場合は自分でそれらのパラメータを加えなければならない。 GAFF が出来るだけパラメータを増やしてくれることを希望したい。表5は作成されたfrcmodである (sustiva.frcmod):
remark goes here |
4つのmissing angle parametersが書かれている。Xleapで後ほどどの原子がこれらに相当するかを検討する。このチュートリアルではAntechamber が示したパラメータは正しいと仮定する。本来は例えばab initio calculations などでこれらのパラメータを検討するのが望ましい。"ATTN: NEEDS REVISION"というコメントの付いたパラメーターがある場合は、antechamberが適したパラメーターを決定出来ないのでシミュレーションを進めるためにはそれらを自分で入力する必要がある。デファオールトとしてantechamberはそれらを0とする。
これまでの作業でLeapでsustiva関係の必要なすべての情報を揃えられた。Leap を実行し、GAFF force field が使用可能かどうかを確認する。traditional AMBER force fields と GAFF を一緒に使えることが可能なので、ここでa fragment of the HIV virus を入力しFF99 force fieldで扱い、sustiva 分子をGAFF force fieldで扱う。このチュートリアルでは、GAFF sustivaを読み込み、truncated octahedral box of TIP3P waterに埋め込む。TIP3P water parameters はFF99 Leap scriptの一部として読み込まれるので、tleapは以下のようにして実行できる(生命環境のコンピュータは、単にtleap enterのみで下記の実行と同じ条件で実行可能)。
$tleap -f oldff/leaprc.ff99SB
tleapが実行されたらGAFF force fieldが利用出来るかを確認する。下記のようにsourceファイルを読み込むと、$AMBERHOME/dat/leap/cmd/のスクリプトにleaprc.gaffが読み込まれたかどうかが示される。
source leaprc.gaff
tLeapのconsoleには下記のテーブルのような記述がある。:
| Welcome to LEaP! (no leaprc in search path) Sourcing: /usr/local/amber10/dat/leap/cmd/oldff/leaprc.ff99SB Log file: ./leap.log Loading parameters: /usr/local/amber10/dat/leap/parm/parm99.dat Reading title: PARM99 for DNA,RNA,AA, organic molecules, TIP3P wat. Polariz.& LP incl.02/04/99 Loading parameters: /usr/local/amber10/dat/leap/parm/frcmod.ff99SB Reading force field modification type file (frcmod) Reading title: Modification/update of parm99.dat (Hornak & Simmerling) Loading library: /usr/local/amber10/dat/leap/lib/all_nucleic94.lib Loading library: /usr/local/amber10/dat/leap/lib/all_amino94.lib Loading library: /usr/local/amber10/dat/leap/lib/all_aminoct94.lib Loading library: /usr/local/amber10/dat/leap/lib/all_aminont94.lib Loading library: /usr/local/amber10/dat/leap/lib/ions94.lib Loading library: /usr/local/amber10/dat/leap/lib/solvents.lib > source leaprc.gaff ----- Source: /usr/local/amber10/dat/leap/cmd/leaprc.gaff ----- Source of /usr/local/amber10/dat/leap/cmd/leaprc.gaff done Log file: ./leap.log Loading parameters: /usr/local/amber10/dat/leap/parm/gaff.dat Reading title: AMBER General Force Field for organic mol., add. info. at the end (June, 2003) > |
つぎにsustiva unit (sustiva.mol2)を読み込む:
SUS = loadmol2 sustiva.mol2
listコマンドを入力するとSUSユニットが下記のように示される (ボールドで示される):
|
> SUS = loadmol2 sustiva.mol2
Loading Mol2 file: ./sustiva.mol2 Reading MOLECULE named SUS > list ACE ALA ARG ASH ASN ASP CALA CARG CASN CASP CCYS CCYX CGLN CGLU CGLY CHCL3BOX CHID CHIE CHIP CHIS CILE CIO CLEU CLYS CMET CPHE CPRO CSER CTHR CTRP CTYR CVAL CYM CYS CYX Cl- Cs+ DA DA3 DA5 DAN DC DC3 DC4 DC5 DCN DG DG3 DG5 DGN DT DT3 DT5 DTN GLH GLN GLU GLY HID HIE HIP HIS HOH IB ILE K+ LEU LYN LYS Li+ MEOHBOX MET MG2 NALA NARG NASN NASP NCYS NCYX NGLN NGLU NGLY NHE NHID NHIE NHIP NHIS NILE NLEU NLYS NMABOX NME NMET NPHE NPRO NSER NTHR NTRP NTYR NVAL Na+ PHE PL3 POL3BOX PRO QSPCFWBOX RA RA3 RA5 RAN RC RC3 RC5 RCN RG RG3 RG5 RGN RU RU3 RU5 RUN Rb+ SER SPC SPCBOX SPCFWBOX SPF SPG SUS T4E THR TIP3PBOX TIP3PFBOX TIP4PBOX TIP4PEWBOX TP3 TP4 TP5 TPF TRP TYR VAL WAT frcmod99SBgaff parm99 |
この時点ではparmchkで得たfrcmodファイルをロードしてはいない。従ってSUSユニットをチェックするとangle type parameterが4つ無いのが分かる。
check SUS
|
> check SUS
Checking 'SUS'.... Checking parameters for unit 'SUS'. Checking for bond parameters. Checking for angle parameters. Could not find angle parameter: ca - c3 - c1 Could not find angle parameter: c1 - c1 - cx Could not find angle parameter: c1 - cx - hc Could not find angle parameter: c1 - cx - cx Could not find angle parameter: c1 - cx - cx There are missing parameters. Unit is OK. |
欠けているangle type parametersはca-c3-c1、 c1-c1-cx、 c1-cx-hc、c1-cx-cxである。これらは、propyl ringとc-cの三重結合である。このことはこれらのシステムが有機分子中ではあまりないので予想されてたことである。前述のfrcmodファイルをこれらの欠けているangle typeのパラメーター入力のためにロードする。
loadamberparams sustiva.frcmod
ここでSUSユニットをチェックすると欠けているパラメーターが無いことが分かる。:
|
> loadamberparams sustiva.frcmod
loading parameters: ./sustiva.frcmod Reading force field modification type file (frcmod) Reading title: remark goes here > check SUS Checking 'SUS'.... Checking parameters for unit 'SUS'. Checking for bond parameters. Checking for angle parameters. Unit is OK. |
ここでsustivaのlibrary( sus.lib)ファイルと、prmtop inpcrd file(sustiva.prmtop, sustiva.inpcrd) を作成する。
saveoff SUS sus.lib
saveamberparm SUS sustiva.prmtop sustiva.inpcrd
tleapのアウトプットファイルには2、3のwarningがあるが、この場合はsustiva: の三角型結合座標によるものなので無視しても問題ない。
|
> saveoff SUS sus.lib
Building topology. Building atom parameters. > > saveamberparm SUS sustiva.prmtop sustiva.inpcrd Checking Unit. Building topology. Building atom parameters. Building bond parameters. Building angle parameters.< br style="font-family: Bitstream Vera Sans Mono;">Building proper torsion parameters. 1-4: angle 7 12 duplicates bond ('triangular' bond) or angle ('square' bond) 1-4: angle 7 9 duplicates bond ('triangular' bond) or angle ('square' bond) < span style="font-family: Bitstream Vera Sans Mono;">1-4: angle 9 12 duplicates bond ('triangular' bond) or angle ('square' bond) Building improper torsion parameters. total 8 improper torsions applied Building H-Bond parameters. Not Marking per-residue atom chain types. Marking per-residue atom chain types. (Residues lacking connect0/connect1 - these don't have chain types marked: res total affected SUS 1 ) (no restraints) > |
tleapのコンソールで全てのコマンドを入力する代わりに上記のコマンドのリストを作りtleap input ファイルとすれば同じことが可能である;input file (tleap.in) :
tleap -f tleap.in
従来のAMBER force fieldはGAFFと共に使用出来るので、HIVの逆転写酵素(RT)のフラグメントをロードし、それをff99SB force fieldで取り扱い、Sustiva分子をGAFF force fieldを用いて取り扱うことが出来る。そのために上記で作成したSustiva library file (sus.lib)が必要である。
RT-Sustiva complexはRCSB protein data bank (pdb code 1FKO)に置かれている。該当のpdbファイルは1FKO.pdbである。.
HIVの逆転写酵素(RT)はp51 と p66のサブユニットで形成されるヘテロダイマーである。それは分子量117kDaの大きなタンパク質である。 このチュートリアルの目的のため、ここではSustivaに近接するp66 subunitのfinger と palm domainを含む切断フラグメントを使用する。切断フラグメントpdbは1FKO_trunc.pdbである。
1FKO pdbファイルをtleapに認識させるために一度Sustiva libraryファイルをロードし、1FKO pdbファイルの残基名EZFをSUSに変更する。Sustiva libraryファイルは1FKO pdbファイル中のSustiva分子と同じ原子名を含んでおり、それ以上の修正は不要である。Sustiva以外のケースではpdbファイルとlibrayファイルが残基名も原子名も同じであることを確認する。修正したpdbファイルは1FKO_trunc_sus.pdbである。pdbファイルをtleapにロードすることが可能である。 まず、tleapを前節と同様に起動する。:
tleap -f oldff/leaprc.ff99SB
>source leaprc.gaff
>loadamberparams sustiva.frcmod
Sustiva library ファイル (sus.lib)をロード後、複合体ファイル1FKO_trunc_sus.pdb.をロードする。
>loadoff sus.lib
>complex = loadpdb 1FKO_trunc_sus.pdb
これでtruncated RT-sustiva complexのtopology と coordinateファイルを作成する準備が整った。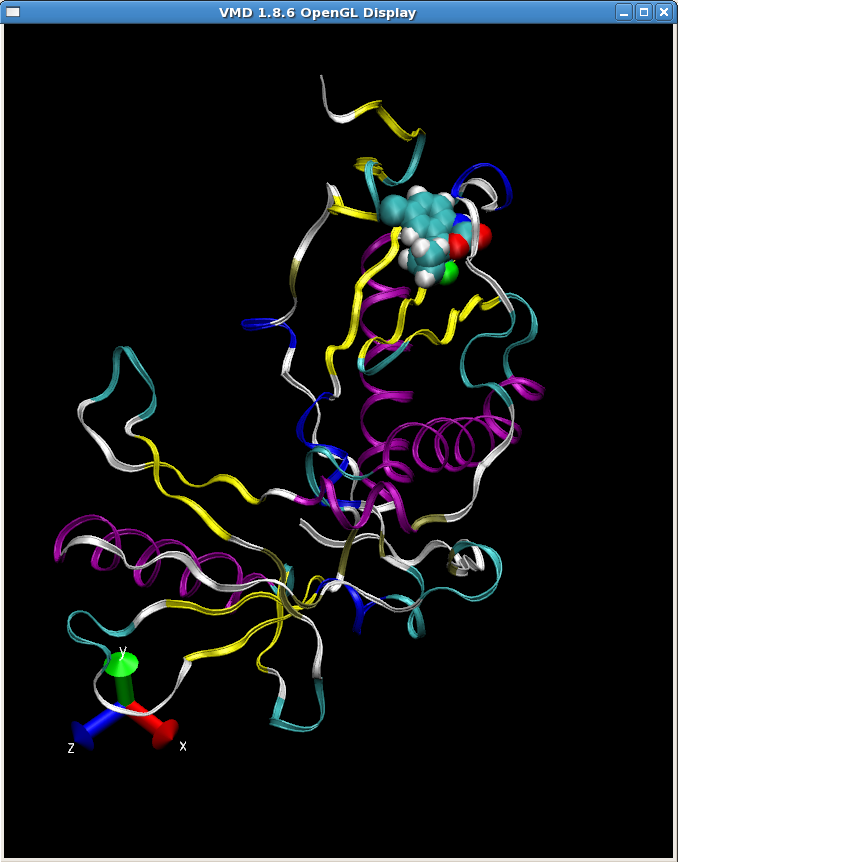
前記と同様にtLeap input ファイル (tleap2.in)を作ればSustiva-RT complexに対する全てのファイルを作成出来る。
tleap -f tleap2.in
RT-Sustiva complexのtopology とcoordinateファイルが作成出来たので、簡単なSustiva-RTのGB simulationが可能である。注意)下記の入力ファイルで実行されるGB MDはこのチュートリアルに対応してごく短いものである。本当の"production"シミュレーションでは普通nsオーダーの実行を行い良好な統計的収束を得るように心がける。
まず存在するかもしれない原子間の不当なコンタクトを取り除くために複合体のエネルギー最小化を行う。そのinputファイルはmin.in である。200ステップのエネルギー最小化 (MAXCYC)を行うが最初50ステップは急勾配法(NCYC)で行い残りは共役法(MAXCYC-NCYC)を実行する。周期的シミュレーションを行うのではないので16Åという十分に大きなカットオッフを用い、静電的正確さを確保して実行する (NTB=0,CUT=16)。 implicit solvent model(連続溶媒モデル?)対してはHawkins, CramerおよびTruhlarのGBモデル(IGB=1)を用いるが詳細についてはAmberマニュアルを参考にする。:|
Initial minimisation of sustiva-RT complex &cntrl imin=1, maxcyc=200, ncyc=50, cut=16, ntb=0, igb=1, &end |
sander -O -i min.in -o 1FKO_sus_min.out -p 1FKO_sus.prmtop -c 1FKO_sus.inpcrd -r 1FKO_sus_min.crd &
必要なら最後のエネルギー最小化構造のpdbをambpdbで作成する。:
ambpdb -p 1FKO_sus.prmtop <1FKO_sus_min.crd > 1FKO_sus_min.pdb
人間の目では異常を見つけ易いので、結果を視覚化することが望ましい。
次のステップはRT-Sustiva complexの温度をあげることである。ためしなので1psに設定してある。理想的にはより長い結果をえることが望ましい。
下記にinputファイルを示した。MDの実行(imin=0)でリスタートではない(irest=0)。この例ではshakeを使っているが、水素の動きが結合エネルギーに影響するかも知れないからである(多分そうでは無いかもしれないが例題であるのでそれを使用した)。shakeを用いない場合は普通より小さいtime stepを使う必要がある。ここではtime stepとして1fsを用い、1000 step実行している。 [1 ps] (dt = 0.001, nstlim=1000, ntc=1)。outputファイルとtrajectroy[mdcrd]は20 stepごとに書き出している(ntpr=20,ntwx=20) 。温度制御は Langevin dynamics approachを用いcollision frequencyは1 ps^-1としている。 0Kで開始し、目的温度を300Kとしている(ntt=3, gamma_ln=1.0, tempi=0.0, temp0=300.0)。input ファイルはeq.inである。
| eq.in Initial MD equilibration &cntrl imin=0, irest=0, nstlim=1000,dt=0.001, ntc=1, ntpr=20, ntwx=20, cut=16, ntb=0, igb=1, ntt=3, gamma_ln=1.0, tempi=0.0, temp0=300.0, &end |
sander -O -i eq.in -o 1FKO_sus_eq.out -p 1FKO_sus.prmtop -c 1FKO_sus_min.crd -r 1FKO_sus_eq.rst -x 1FKO_sus_eq.mdcrd &
温度上昇のheating trajectory と restart coordinateはそれぞれ 1FKO_sus_eq.mdcrd と 1FKO_sus_eq.rstに保存される。 sustiva-RT complexのエネルギーは最小化され、そのあと熱せられた。その300Kでのスナップショットを見ることが出来る。この構造はつぎの平衡化の出発構造として用いることが可能である。1Wang, J., Wolf, R.M., Caldwell, J.W., Kollman, P.A., Case, D.A. "Development and Testing of a General Amber Force Field", J. Comp. Chem., 2004, 25, 1157 - 1173.
2Bayly, C.I., Cieplak, P., Cornell, W.D., Kollman, P.A. "A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges : The RESP Model", J. Phys. Chem, 1993, 10269-10280.
3Jakalian, A., Bush, B.L., Jack, B.D., Bayly, C.I., "Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method.", J. Comp. Chem., 2000, 21, 132-146.