前記したように、分子動力学 trajectory ファイルの分析は高度に標準化された作業です。全ての分析を網羅する方法を予測し、簡潔にまとめたパッケージで実行することは難しい(それが出来ればすばらしいことは当然ではあるけれど。)前のセクションで既に trajectoryファイルを使い RMSD を計算する方法を見た。
I. Determination of the initial relaxation time by trace-back RMSD analysis
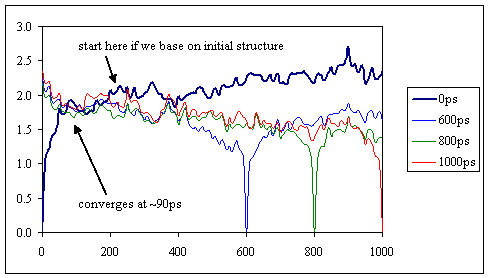
このセクションでは equilibration 問題点を取り扱う。最初に、経験に裏付けられた推測と 100psのシステム平衡化を行う。次に、 potential energy と rmsdを検討し、 trajectory が安定でありデータ収集を初めても良いかを決定する。 J. Chem. Phys. Vol 109(23), 10115, の論文でStella等は、初期構造からの root mean square deviation による初期 relaxation timeの決定は、しばしば真の値を過大評価しその結果、高価値のシミュレーションデータを無駄にすることになると述べている。彼らは初期緩和過程にtrajectoryがどの程度依存するかを決めるより良いアプローチを提案しており、初期に捨てられたデータの回復法を提供している。彼らのアプローチは後期構造をリフェレンスにしてrmsd値を計算し、それを初期構造を使って計算したrmsd値と比較することを含んでいる。カーブを時間に沿って逆にたどり、trajectoryがステイショナリ状態に達するポイントをより良く決めることが出来る。この方法を 最初の 100-ps 定圧-定温平衡trajectoryを含む1 ns trajectory の検討に応用する。既に、単一のcrdファイルを trajectory ファイルに結合し、再イメージ化してファイルを用意してある。ファイルサイズを小さくするために水とカウンターイオンを trajectory から取り除いてある。もし読者自身でこの作業をするとすれば、tleapで新しいtopology ファイルを作りtrajectoryから不要部分を取り除く必要がある。これには3ステップの作業が必要である。
1. 初期 topology とrestartファイルを用いpdbファイルを作成する。2. wt1mg_water.pdb ファイルから水分子、Cl- イオンを取り除く。(Macではエディタmiを用いる)
3. tleap を用いて、水を取り除いたpdbを入力し、新しい topology ファイルを保存する。訳注)普通の環境ではこれではうまく行かない場合
>source leaprc.ff99SB
>loadoff MG.off
> mol = loadPdb wt1mg_water.pdb
>saveAmberParm mol wt1mg_dry.parm7 wt1mg_dry.rst
とする。
trajectories を cleaning up (この意味は水を取り除き中心を1-152残基に置くということ)するptraj インプットファイルは以下のようになっている:ptraj_cleaning.inとして保存する。
これらのファイルはダウンロードページからダウンロード出来る。
少しすっきりしないシェルスクリプトだが次記のようにしてこのファイルを作った。
(wt1mg_eq.crd とwt1mg_md.crdファイルはダウンロードにない。equibrationとproductionで示した実行ファイルにより作成する。 このマニュアルではファイル名にdryが無い場合すべてwatファイルである。)
### take snapshots at 100ps generate reference files
cat << EOF > ptraj.in
trajin wt1mg_dry.crd 1 100 10
trajout wt1mg_ref restart
go
EOF
ptraj wt1mg_dry.parm7 ptraj.in
### calculate the rmsd for each reference structure
foreach ref (`ls wt1mg_ref*`)
set ofile=$ref.rms
cat << EOF > rms.ptraj
trajin wt1mg_dry.crd 1 100
reference $ref
rms reference out $ofile :1-154
go
EOF
ptraj wt1mg_dry.parm7 rms.ptraj
end
### clean up the files and merge them into a single file
cp wt1mg_ref.2.rms rms1
set i=10
while ($i <= 100)
cut -b12-20 wt1mg_ref.$i.rms > tmp
paste rms1 tmp > rms
cp rms rms1
@ i=$i + 10
end
rm *.rms rms1 tmp
mv rms relax.rms
上記スクリプトを実行すると出力として "relax.rms" を得る。下記図より結論を出す。 ;-)
II. B-factor calculation
前記以外の一般的に行われている検討は MD trajectory から計算してB- factorと、結晶学によるB-factorの比較である。これはptrajを用いれば簡単に行われる。 そのptraj インプットファイルは、下記に示すptraj_bfactor.inである。
ptraj input file は:
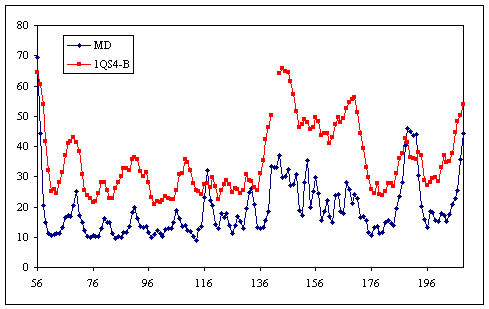
このptraj でそれぞれの C-alpha 原子に対する B-factor が得られる。 1QS4 chain B pdbファイルの実験 B-factorと上記結果をプロットした (これは最初用いたモデルの構造である)。 2〜3の重要な残基を除いてMDシミュレーションによるB-factorは 結晶学B-factor より遥かに小さいことが分かる。このことは 1-ns が結晶構造と比べると十分なサンプリングでないことから予測される。
III. Visualization
最後に、MDプロジェクトは映像で見れないのか?
AMBER フォーマットの trajectory を再生出来るフリーのプログラムがいくつか存在している (PyMol, VMD, その他)。 VMD は AMBER trajectory を映像として再生する非常に良いプログラムで、データをインターラクティブに扱う使いやすいツールを備えている。 1-ns trajectory を楽しめる短い映像を作成しておいた。どんな見識をMDシミュレーションから得られた?金の採掘のようにそこには全てが眠っており新しい発見がそこに待っている。