tleap を用いて当該分子にwater boxを加え、モデルシステムにカウンターイオンを加える。
上記のように, 水のボックスを加えるコマンドは "solvateBox"である。(ただし新しいamber 少なくともamber12ではWATBOX216がTIP3PBOXになる。 いくつかの水を加える方法はあるが、ここでは最も直接的な方法を採用する。 WATERBOX216 (TIP3PBOX)は TIP3P water の平衡化済みboxである。数字の10はオングストローム単位で示されたタンパク質とboxのedgeの緩衝距離である。使用するバッファーのサイズはその都度考慮するする必要がある。もし大きすぎるバッファーを指定すると意味の無い水に対する余分な計算時間を必要とされる。しかし、小さすぎると該当分子がコンフォメーションチェンジし、その一部がwater boxからはみ出すかもしれない。もし実験条件になるべく近いシミュレーションを行いたい場合で、あらかじめシステムの濃度が分かる場合はその情報から必要なwater boxのサイズを知ることが可能でありそれを明確に設定することが出来る。そうでなければ10を初期値として設定すれば十分である。次にカウンターイオンを加える。 "addions" コマンドを使う前に、本システムが負に荷電しているか正に荷電しているかを明らかにする。もし正に荷電しておれば負のイオンCl- を加え、負に荷電していればNa+ イオンをカウンターイオンとして加える。システムのチャージを決めるには "charge"コマンドを使う。:
本システムでは正に荷電しているのでCl- イオンをを用い荷電をバランスさせる。 AMBER は実際にイオンを付加する2つのアルゴリズムを提供している。 "addions" コマンドで実行されている最初の方法は、単純に溶質の周囲にグリッドを描きエネルギーが最小になるようにグリッドポイントにイオンを置いている。 この方法はイオンを置く位置にある水分子を無視しており、もしイオンを置く位置に水分子が存在すると水分子は取り除かれイオンに置き換えられる。このアルゴリズムを用いると、我々の意図とは別に、 Mg2+の隣に Cl-イオンを置くことになる。 第二の方法は "addions2" であり "addions" コマンドとほぼ同じであるが、溶質分子を溶媒分子と同様に扱う。この例題では "addions2" を用い、 Cl- イオンは扱う分子から幾分離れて置かれ、人為的にシステムを歪ますことなくチャージを与える。数字の0はシステム全体のチャージを0にするためである。
これで全てでinput filesとして出力して保存する。
tleapを終了する。
-----------------------------------------------------------------------------------------------------------------
http://masa-cbl.hatenadiary.jp/entry/20121104/1352014396
によると
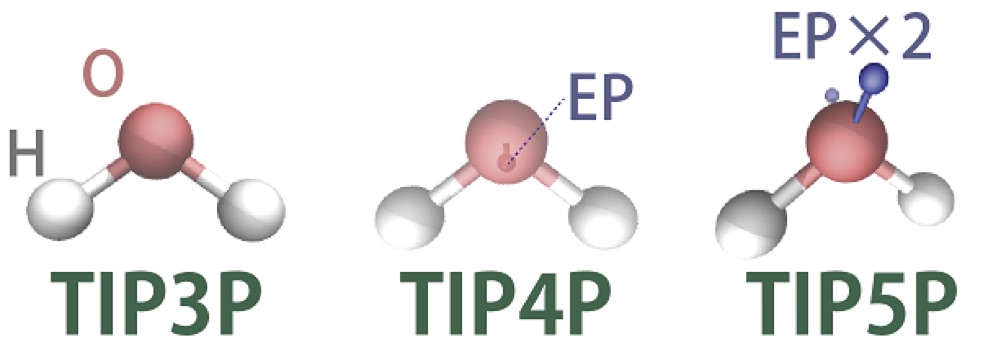
分子動力学計算で使われる水モデルの構造と使用頻度
分子シミュレーション:
分子動力学(MD)計算において最も頻繁に使われる分子として水分子H2Oがあります。
水そのものの性質を調べることはもちろん、それ以外にも水溶媒中の生体高分子等の計算にも水分子は必要であり、そのモデルの正確性は結果に大きく影響し重要です。
3相互作用点水モデル:
現在最もよく使われる水のモデルは各O原子、H原子2原子それぞれに1つずつ相互作用点を配置し剛体として扱う3相互作用点剛体モデルの一種であるTIP3Pモデルで、AMBERでもデフォルトだとこのTIP3Pモデルが使われます。
ただしTIP3Pモデルには拡散係数が実験値より2倍以上大きいという欠点があります。
他にも代表的な3相互作用点剛体モデルとしてSPC, SPC/Eモデルが、また使用頻度は低いですが剛体として扱わずOH間の振動等の分子内自由度を付与したモデルとしてSPC/Fモデルがあります。
と書いてある。詳細はそのサイトを参考に。)
下記にしめすようにこのチュートリアルを書いてる時点の Pentium IV PC ではこの計算に990秒かかっていたので気長に待つ必要があったが、現在のi7ではせいぜい100秒程度なのですぐに終わる。結果をアウトプットファイルとして保存する。そしてtleapコマンドを閉じる。cp12ではチャージがマイナスなのでNa+をカウンターイオンに用いる。cp12のtleapインプットファイルは: