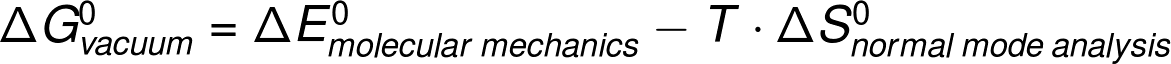
(Note: のチュートリアルでは MM-PBSA (molecular mechanics Poisson-Boltzmann surface area)法を二つのタンパク質の結合エネルギーを計算するために用いる。)
Copyright Ross Walker 2006
AMBER ADVANCED TUTORIALS
TUTORIAL 3
MM-PBSA
Perl Version By
Ross Walker & Thomas Steinbrecher
Python Version By Dwight McGee, Bill Miller III, & Jason Swails
このチュートリアルでは MM-PBSA (molecular mechanics Poisson-Boltzmann surface area)法を二つのタンパク質の結合エネルギーを計算するために用いる。
MM-PBSA法とその相補的 MM-GBSA法の総合的目的は、溶媒和された2分子が結合と解離状態にある時、または一つの分子が異なる状態にある時の自由エネルギー差を計算することである。
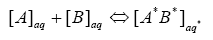
理想的には、下図に示すように、結合の自由エネルギーを直接計算したい。:
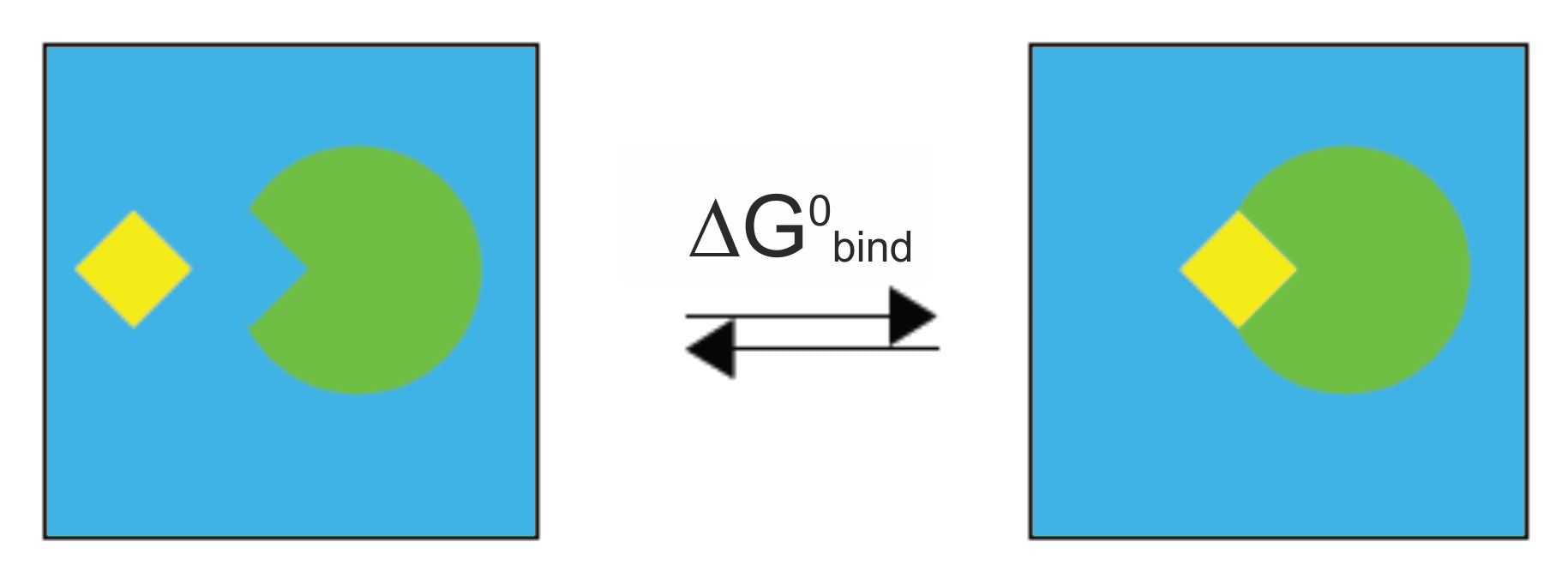
しかし、溶媒和の状態でのシミュレーションは、エネルギーの寄与の大部分は溶媒-溶媒相互作用から生じ、総エネルギーの変動は結合エネルギーよりもワンオーダー大きい。したがって、計算が収束するには時間がかかるので、以下の熱力学サイクルに応じて計算を分割し効率的に実行する。:
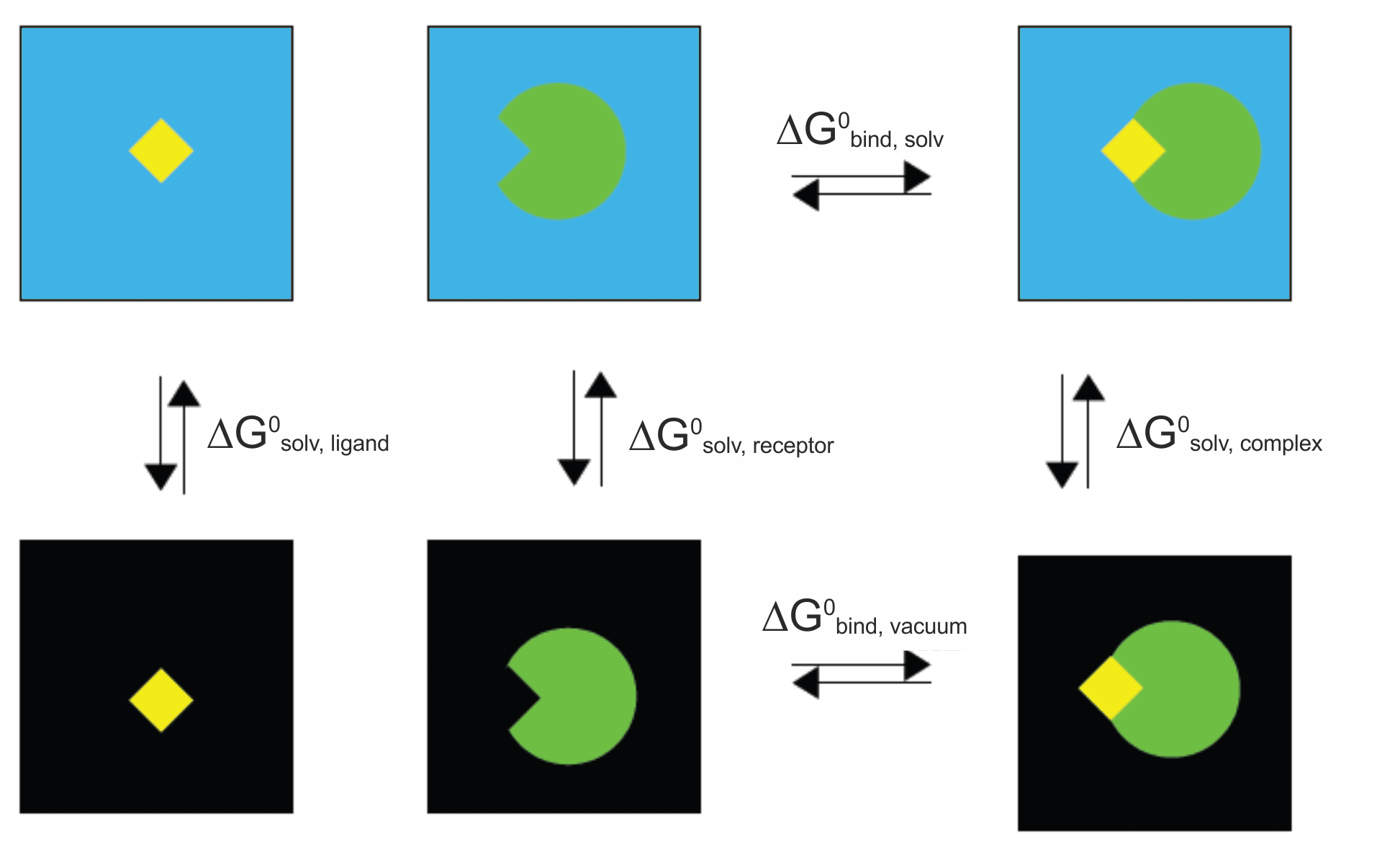
このダイアグラムから明らかなように自由エネルギーdelta-Gbind,solv下式により計算できる。:
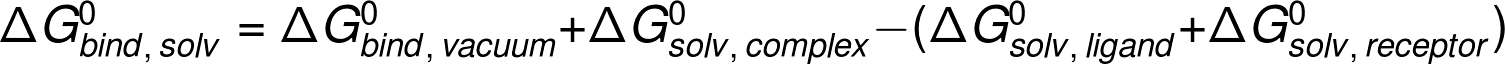
MM-PBSAアプローチでは、上記の結合自由エネルギーへのそれぞれの寄与は、いくつかの方法で計算される。:
溶媒和自由エネルギーは、次の3つの状態の直線化ポアソン・ボルツマン方程式またはGeneralized Born方程式を解き(これは溶媒和自由エネルギーに対する静電寄与を与える)、疎水性寄与に関する経験的な項を追加することにより計算される。:
delta-Gvacuum は、受容体とリガンドとの平均相互作用エネルギーを計算し、必要か望ましい場合、結合エントロピー変化も考慮することによって得られる。:
エントロピーの寄与は、三分子の通常モード分析を行うことにより求めることができるが、二つの配位子が同一のタンパク質に結合するような同様のエントロピーの状態の比較のみ必要な場合、実際にはエントロピーの寄与を無視することができる。この理由は、通常モード分析計算はコンピューター的にはハイコストであり、結果に大きい不確実性を導入する大きなマージンの誤差を持つためである。
受容体とリガンドの相互作用の平均エネルギーは、通常、平衡化した分子動力学(MD)シミュレーションで収集される無相関スナップショットの集合に演算を行うことによって得られる。
このチュートリアルでは、自動的に、タンパク質-タンパク質複合体(RASとRAF)とタンパク質-リガンド複合体(エストロゲン受容体およびラロキシフェン)の結合自由エネルギーをMM-GBSAとMM-PBSAシリアルとパラレルのメソッドの両方を使用して推定する必要なすべての手順を実行する。これらのメソッドはAmberとAmberToolsに含まれているMM/P(GB)SAスクリプトの使用により実行できる。さらに、スクリプトを使用して、アラニンスキャニングおよびノーマルモードのエントロピーの計算を実行する。原理的には、上述の結合自由エネルギーの計算は、複合体と個々のタンパク質の独立したMDシミュレーションが必要である。しかし、典型的には、結合時に有意なコンフォメーション変化が無いので、3つすべての種についてのスナップショットは、単一のtrajectoryから得ることができるという近似を行う。これは、「単一のtrajectoryアプローチ」であり、このチュートリアルで使用する。
Section 1 : 出発構造を構築し、平衡化したシステムを取得するために、シミュレーションを実行する。
Section 2 : productionシミュレーションを実行し、スナップショットensembleを得る。
Section 3 : 結合自由エネルギーを計算し、その結果(異なるバージョンMM/PBSA間で、チュートリアルは分岐する)を分析。
(Note:これらのチュートリアルは、妥当な時間内に、簡単なワークステーション上で実行することができ、シミュレーションを実行するためにAMBERソフトウェアスイートを使用する方法の例を提供することを意図している。特定のアプリケーション領域のための最適なパラメータまたは方法を提供していない。) Copyright Ross Walker 2006