チュートリアルA24
第3章:線形相互作用エネルギー解析
FEWはで詳細に線形相互作用エネルギー(LIE)アプローチに従って、結合自由エネルギーの計算を行うための機能を提供する[3,
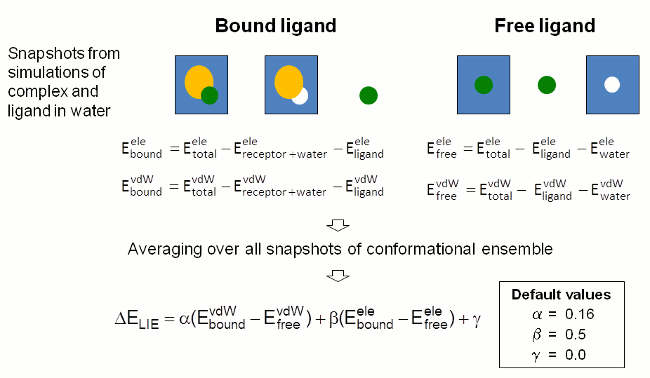
4]。このアプローチで結合自由エネルギーは、複合体形成時にリガンドと環境との間の静電的(ELE)及びファンデルワールス(VDW)の相互作用エネルギーの変化から推定する。相互作用エネルギーの寄与は、溶媒中の遊離型および複合体結合型リガンドのMDシミュレーションスナップショットにより、溶媒和システムの総エネルギーから個々のコンポーネントのエネルギーを減算することにより(図4)求める。エネルギーは、sanderを用いた一点計算によって決定する。個々のスナップショット対して計算された相互作用エネルギーは、ユーザにより評価のために選択された立体配座のアンサンブル全体にわたって平均化される。
結合自由エネルギーは、図中の重み係数によってスケーリをされたvdWと静電相互作用エネルギーの差の総和(図4の最後の式を参照)として推定する。FEWは、研究対象システムに対するリガンドの結合自由エネルギーの概算を与えるために確立された重み係数を用いる。
| |
| 図 4: LIE アプローチによって自由エネルギーをリガンド結合の推定のたのFEWに用いられる手順の概略図。 |
FEWによるLIEのセットアップの前提条件は、FEWのMDセットアップ機能を使用して生成された溶液中のリガンドと複合体の軌跡の存在である。このような軌跡をMM-PBSA計算に対する3-軌道のセットアップを用いるチュートリアルsection 1で作成したので、すべての必要な情報はすでに入手可能である。LIE分析を用意するためのコマンド・ファイルは、/home/user/tutorial/cfiles directoryにあるlie_am1で提供している。
ヒント: LIE計算のみを実施するなら、traj=3 でMDシミュレーションを用意し、FEW プログラムでのcallでMMPBSAを使わずにLIEを使う。この方法ではMDシミュレーションのみが複合体とリガンドに対して用意されており、LIEに対しては過剰な受容体のシミュレーションは用意されない。| lie_am1 |
| @LIEW ################################################################################ # Command file for LIE calculations based on trajectories # generated by molecular dynamics simulations previously. ################################################################################ # Location and features of input and output directories / file(s) # # lig_struct_path: Folder containing the ligand input file(s) # output_path: Basis directory in which all setup and analysis folders will # be generated. The directory needs to be identical with the # 'output_path' directory used for setup of the MD simulations. lig_struct_path /home/user/tutorial/structs output_path /home/user/tutorial # Receptor features # water_in_rec: Water present in receptor PDB structure # used for setup of MD simulations water_in_rec 1 ################################################################################ # General Parameters for LIE calculation setup # # lie_calc: Request setup of LIE calculations # charge_method: Charge method used for MD, either "resp" or "am1" # no_of_rec_residues: Number of residues in receptor structure # additional_library: If an additional library file is required, e.g. for # non-standard residues present in the receptor structure, # this file must be specified here. # additional_frcmod: If additional parameters are needed, e.g. for describing # non-standard residues present in the receptor structure, # a parameter file should be provided here. # lie_executable: Location of LIE calculation script provided with FEW. # If no script is specified, it is assumed that the LIE # program is located in the default directory. # lie_batch_template: Template batch file for LIE calculation # lie_batch_path: Path to regard as basis path for setup of batch files # lie_calc 1 charge_method am1 no_of_rec_residues 290 additional_library /home/user/tutorial/input_info/CA.lib additional_frcmod lie_executable lie_batch_template /home/user/tutorial/input_info/lie.sge lie_batch_path # ################################################################################ # Parameters for coordinate extraction # # image_trajectories: If set to "1" solutes of the specified trajectories # will be imaged to the origin before coordinates are # extracted. Please regard that this may require a large # amount of additional disc space. # trajectory_files: Trajectory files to regard. The path will be determined # automatically. Specify 'all' to regard all trajectories # produced in the production phase of the MD simulations. # This ensures consistent snapshot numbering. # Subsets of snapshots will be generated according to the # parameters first_lie_snapshot, last_lie_snapshot, and # offset_lie_snapshots. If only a subset of the trajectories # from MD shall be used, the individual files must be # specified as 'trajectory_file # entry per line. # ATTENTION: The trajectory file(s) must exist for both the # simulation of the complex and the simulation of # the free ligand in solution. # snaps_per_trajectory: Number of snapshots stored in each trajectory. # # trajectory_files md_prod_001.mdcrd.gz trajectory_files md_prod_002.mdcrd.gz # snaps_per_trajectory 50 ################################################################################ # LIE Analysis # # calc_sasa: Request calculation of change in solvent accessible surface area # first_lie_snapshot: No. of snapshot from which the LIE calculation # shall be started # last_lie_snapshot: No. of last snapshot that shall be regarded in the # LIE calculation # offset_lie_snapshots: Offset between snapshots that shall be regarded # in the LIE calculation # sander_executable: Sander executable that shall be used for the LIE # calculation, if this differs from the default one # in $AMBERHOME/bin # parallel_lie_call: Call for running sander in parallel, e.g.: # mpirun -np 2 -machinefile $PBS_NODEFILE # Prerequisite: Parallel version of sander installed # delete_lie_trajectories: Delete trajectory files created for LIE analysis # directly after energy calculation # calc_sasa 0 first_lie_snapshot 81 last_lie_snapshot 100 offset_lie_snapshots 1 sander_executable parallel_lie_call mpirun -np 8 delete_lie_trajectories 0 |
LIE分析のコマンド・ファイルは、次の部分から成り立つ:
1.入力および出力ディレクトリ/ファイル(複数可)の場所:
ここで入力/出力の特定のパラメータが定義される。スクリプトのこの部分は、以前に示した別のコマンドファイルの該当部分と同じである。(参照例 section 1)
2. LIE計算セットアップのための一般的なパラメータ : 3. 座標 抽出 のための パラメーター: 4. LIE 分析の パラメータ:
FEWの実行: 出力: /home/user/tutorial/lie_am1/qsub_LIE.sh ヒント:
実験的な結合親和性が知られているデータセットに対して線形フィットで最適重み係数を決定したい場合、 FEWで作られたextract_LIEenergies.plスクリプトを使用して、データセットのすべての結果ファイルからすべての静電的およびVDW相互作用エネルギーの違い(ならびに溶媒接近可能表面積)を抽出することができる。詳細ついてはFEWのマニュアルを参照しなさい。 SECTION 2: MM-PB(GB)SA calculations Copyright:
Nadine Homeyer 2014
LIE 分析はam1チャージがリガンドに対して使用されているMDシミュレーションに基づいて設定されている。受容体の残基290に構造的に結合したカルシウムイオンに対しては、追加のライブラリファイルをロードする。 実行可能LIE.pl
はデフォルトの場所にあり、LIEの計算はbasic input/output directoryを出力ディレクトリとしてセットアップされる。バッチファイル設定のテンプレートとしてサーバーはlie_batch_templateにあるファイルを指定した。このテンプレートファイルでは、計算準備セクション前の設定でLIE計算が行われるコンピューティング環境に適応する必要がある。
コマンドファイルのこの部分は、このチュートリアルのMM-PB(GB)SAの計算のため書かれたsection 2のコマンド・ファイルのスナップショット抽出の特定の部分に似ている。
軌道のイメージングの前に座標抽出が要求される。MD production時に生成されたすべての軌道が規定されている。これは、スナップショット座標の正確な同定を保証するために必要とされる。LIE計算で考慮しなければならないスナップショットは、下記のsection 4にある各軌道のファイルに存在するスナップショットの数、
first_lie_snapshot、
last_lie_snapshot、 及び
offset_lie_snapshots
に基づいて選択される。
LIE の分析は、
offset
が
1の81
から
100までのスナップショットに基づかなければならない。
FEWは、結合および遊離状態間のリガンドの溶媒近接可能表面積の差を計算する可能性を提供している、なぜならこの値はリガンド特定係数γを決定するために使用されている(図4の最後の方程式参照)。
[8,
9,
10]。 しかし、ここでは、このオプションが選択されていない (calc_sasa=0)
。 for sanderに対するコンピューティング環境の特定のparallel_lie_callが与えられている。LIE分析は結果的に$AMBERHOME/bin/sander.MPI.の下にある並列バージョンのサンダーで実行される。オリジナルのMD軌道からLIE分析のために生成された軌道は、明示的に水分子を含み、ディスクスペースを大量に使うので、1リガンドに対するLIE計算が完了した直後にそれらを削除する機能をFEWは持つ。このオプションは、ここではオンにされなかった (delete_lie_trajectories=0)。
コンピューティング環境固有の設定がされ、コマンドファイル内に正しくbasic input/output pathが指定されておれば、LIEの計算はFEWを用いて調製できる。
FEWが正常終了した後、/home/user/tutorialディレクトリには新しいフォルダlie_am1が含まれ 図5に示すサブ構造を持つ。Please note図5に紫で描かれたエネルギー計算出力フォルダがまだ存在しない。それらはLIE計算中に作成される。
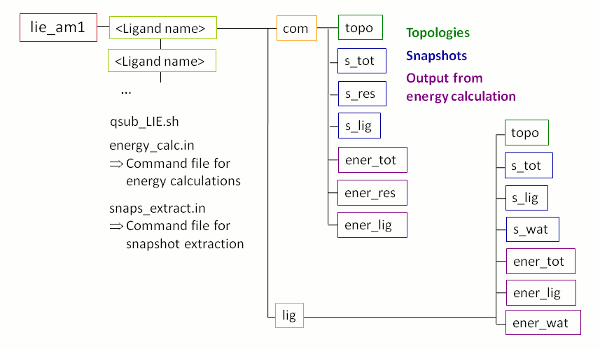
図 5:
FEW 使用してLIE分析に作成したフォルダのサブ構造の模式図
LIE計算の開始:
LIE の計算は、lie_am1にある
シェルスクリプトqsub_LIE.sh
を実行することで起動することができる。
LIEの結果:
LIE 計算の最終結果は、/home/user/tutorial/lie_am1
ディレクトリにある特定のリガンドのサブフォルダに見つけることができる。
ファイルLIE_s<S>_<T>_<O>.txt
は
<S>= first_lie_snapshot,
<T>
= last_lie_snapshot,
<O>
= offset_lie_snapshots, すなわち、 このチュートリアルでファイル
LIE_s81_100_1.txt
は、
複合体結合と遊離のリガンドに対する計算された平均静電的およびvdW相互作用エネルギー(そして溶媒近接可能表面面積)、とその差を示す。
また、図4に示したスケーリングファクタを使用して、結合自由エネルギーの推定値が与えられる。
Result file LIE_s81_100_1.txt
of ligand L51c
Energy contributions:
Mean energy Standard deviation Standard error
Complex non-bonded VDW: -76.550390 3.354497 0.750088
Complex non-bonded ELE: -49.058070 7.613551 1.702442
Ligand non-bonded VDW: -43.399665 3.762162 0.841245
Ligand non-bonded ELE: -56.828235 6.755975 1.510682
================================================================================
Final LIE results:
Differences in interaction energies and SASA:
Delta electrostatic energy (dE-ele): 7.770165 kcal/mol
Delta van der Waals energy (dE-vdW): -33.150725 kcal/mol
________________________________________________________________________________
Estimated binding free energy:
ATTENTION: Please note that the estimate of the binding free energy (dG-bind)
provided below was calculated according to the equation
dG-bind = alpha * dE-vdW + beta * dE-ele
with fixed coefficients of alpha=0.16 and beta=0.5.
We strongly recommend to thoroughly investigate whether using other
coefficients could be beneficial in the specific case.
Binding energy estimate (dG-bind): -1.419034 kcal/mol