Tutorial A24
セクション2:MM-PB(GB)SAの計算
FEWは、MM-PBSAとMM-GBSAのアプローチに従って、平均的な溶媒環境をパラメータとして近似的に取り扱う溶媒分子力学計算を準備することができる。MM-PB(GB)SA計算設定の前提条件は、MDセットアップ機能により生成されたMDシミュレーションフォルダ内のMD軌道の存在である(このチュートリアルsteps 1A & 1Bを参照する)。 FEWは、1-軌道と3-軌道アプローチの両方に対して、MM-PB(GB)SAの計算を準備することができる。ここでは、このチュートリアル(Table1)のセクション1で3つのファクターXa阻害剤に対して調製されたMDシミュレーション二種のいずれかの方法を適用する。
MM-PB(GB)SA法の基本的な概要については文献とTutorialA3を参考にする。Tutorial A3で説明した結合自由エネルギーΔGbind,solvの算出の基本原則は ここで行った計算にも適用される。しかし、結合によってリガンドの立体配置エントロピーは有意に変化しないと仮定されるので、結合のエントロピーへの寄与は考慮しない。ここでは効果的な結合エネルギー ΔGeffectiveとは、FEWを用いてMM-PB(GB)SAの計算の設定によって計算された結合エネルギーを指す。
注意<: ここに行ったサンプルMM-PB(GB)SAの計算は、section 1の2ナノ秒で作製したMDシミュレーションのスナップショットに基づいている。従って、計算に使用されるスナップショットが完全に平衡化構造を表すことは期待できない。さらに、スナップショットの数が少ないので予測の不確実性が増す。ここで示したMM-PB(GB)SAの計算は、FEWの機能性を実証するための例として見なされる。 どのリアルな研究のでも、完全にMM-PB(GB)SAの計算のための代表的な構造アンサンブルを識別するために、MDの軌跡を完全に解析することを勧める。何故ならただこのようなアンサンブルに基づいたMM-PB(GB)SA計算のみが正確な結合エネルギーの予測につながる。平衡状態の研究のためのいくつかの基本的な分析はチュートリアルA3 section 2で見つけることができる。
A: 3-軌道アプローチ
section 1の3-軌道アプローチに従ってMDシミュレーションを実行しているので、3-軌道FEWコマンドファイルana_am1_3trj_pb0_gb2を使用して、直接MM-GBSAのセットアップを開始することができる。| mmpbsa_am1_3trj_pb0_gb2 |
| @WAMM ################################################################################ # Command file for MM-PBSA / MM-GBSA calculations based on trajectories # generated by molecular dynamics simulations previously. ################################################################################ # Location and features of input and output directories / file(s) # # lig_struct_path: Folder containing the ligand input file(s) # output_path: Basis directory in which all setup and analysis folders will # be generated. The directory needs to be identical with the # 'output_path' directory used for setup of the MD simulations. lig_struct_path /home/user/tutorial/structs output_path /home/user/tutorial # Receptor features # water_in_rec: Water present in receptor PDB structure # used for setup of MD simulations water_in_rec 1 ################################################################################ # General Parameters for MM-PBSA / MM-GBSA calculation setup # # mmpbsa_calc: Setup MM-PBSA / MM-GBSA calculations # 1_or_3_traj: "1" or "3" trajectory approach # charge_method: Charge method used for MD, either "resp" or "am1" # additional_library: If an additional library file is required, e.g. for # non-standard residues present in the receptor structure, # this file must be specified here. # additional_frcmod: If additional parameters are needed, e.g. for describing # non-standard residues present in the receptor structure, # a parameter file should be provided here. # mmpbsa_pl: Path to mm_pbsa.pl script mmpbsa_calc 1 1_or_3_traj 3 charge_method am1 additional_library /home/user/tutorial/input_info/CA.lib add_frcmod mmpbsa_pl $AMBERHOME/bin/mm_pbsa.pl ################################################################################ # Parameters for coordinate (snapshot) extraction # # extract_snapshots: Request coordinate (snapshot) extraction # snap_extract_template: Template file for extraction of coordinates from # trajectory, i.e. input-file for mm_pbsa.pl; only # required if non-standard input-file shall be used. # image_trajectories: If set to "1" solutes of the specified trajectories # will be imaged to the origin before coordinates are # extracted. Please regard that this may require a large # amount of additional disc space. # trajectory_files: Trajectory files to regard. The path will be determined # automatically. Specify 'all' to regard all trajectories # files produced in MD. This ensures consistent snapshot # numbering. Subsets of snapshots will be generated according # to the parameters first_snapshot, last_snapshot, and # offset_snapshots. If only a subset of the available MD # trajectories shall be used, the individual files must be # specified as 'trajectory_files # one entry per line. # first_snapshot: First structure that shall be extracted from trajectory # last_snapshot: Last structure that shall be extracted from trajectory # offset_snapshots: Frequency of structure extraction # image_trajectories 1 # trajectory_files all # first_snapshot 1 last_snapshot 100 offset_snapshots 1 ################################################################################ # MM-PBSA / MM-GBSA Analysis # mmpbsa_template: Template file for MM-PBSA / MM-GBSA analysis - File used # as input-file for mm_pbsa.pl; only required if non-standard # file shall be used. # PB: If not zero PB calculation will be performed # Options: "0" -> No PB # "1" -> PB with calculation of the non-polar part of the # solvation free energy using the Method developed by # Tan et al. (J. Phys. Chem. B, 2007, 111, 12263-12274). # This method can only be run in combination with GB=1 # or GB=0. # "2" -> Hybrid model developed by H. Gohlke and A. Metz # with IVCAP=5 and CUTCAP=50 # "3" -> PB with MS=1 and Parse radii # "4" -> PB with MS=1 and mbondi radii. This method can only # be combined with GB=1 or GB=0. # GB: If not zero GB calculation will be performed # Options: "0" -> No GB # "1", "2", "5" -> GB analysis according to 'igb' (see manual) # # decomposition: If larger 0 energy decomposition of specified type is # performed. Options: 1-4 - See Amber manual for decomposition # type options. Decomposition only works with PB=4 and GB=1. # SASA is calculated by the ICOSA method. # no_of_rec_residues: Number of residues in the receptor structure # # total_no_of_intervals: Total number of intervals to analyze. # The total_no_of_intervals needs to be consistent with # the number of 'first_PB_snapshot', 'last_PB_snapshot', # and 'offset_PB_snapshots' definitions below. Setting # total_no_of_intervals to a value larger than 1, is # usually only necessary if snapshots with different # offsets shall be analyzed. # first_PB_snapshot: Structure to start analysis with # last_PB_snapshot: Last structure to regard in analysis # offset_PB_snapshots: Specification of offset between structures that shall # be regarded in the MM-PBSA calculation # # mmpbsa_batch_template: Batch script template for MM-PBSA calculation # mmpbsa_batch_path: Optional, path to regard as basis path for batch script # setup, in case it differs from mmpbsa_template PB 0 GB 2 # decomposition 0 no_of_rec_residues 290 # total_no_of_intervals 1 first_PB_snapshot 51 last_PB_snapshot 100 offset_PB_snapshots 1 # mmpbsa_batch_template /home/user/tutorial/input_info/MMPBSA.sge mmpbsa_batch_path /home/user/tutorial # mmpbsa_sander_exe parallel_mmpbsa_calc 1 |
1.
入力 と 出力 ディレクトリ / ファイル(複数可)の位置(s):
このチュートリアルsection 1で説明した入力/出力情報の部分と同じである
2. MM-PBSA/MM-GBSAの一般的なパラメータ計算セットアップ:
MM-GBSA計算は、AM1-BCC法により決めたリガンド原子電荷を用いて生成された3-軌道法に従って準備されている(1_or_3_traj=3)。水を含まないトポロジーファイルのセットアップのためにカルシウムイオンの追加のライブラリファイルを再び使用し、MM-GBSA分析は、AMBERのローカルインストールディレクトリのbinにあるmm_pbsa.plスクリプトで行われる。
3. 座標 (スナップショット) 抽出するためのパラメータ:
映像化の後このュートリアルで作成したsection 1にある2 ns座標からスナップショット間に(offset_snapshots=1)のオフセットなく抽出されたスナプショット1〜100が要求される。
4.MM-PBSA / MM-GBSA 分析:
AMBER12のigb=2に対応したOnufriev et al. 2004[6]の方法に従ったMM-GBSA計算が必要である。
受容体として構造的に結合されたイオンは、ここで受容体の一部とみなされるので受容体残基の総数は最大で290となる。計算は、スナップショット51から100までオフセット1で実行する。
計算パラメーターはmm_pbsa.plがディレクトリとして
basic input/output directory/home/user/tutorial
ディレクトリを使いシリアルに設定される。
バッチスクリプトMMPBSA.sge
をジョブサブミッションのテンプレートバッチファイルとして使う。
使用するコンピューターの構造環境に従ってこのスリプトのPrepare
calculationのヘッダーセクションを修正する。
$FEW/FEW.pl MMGBSA /home/user/tutorial/cfiles/mmpbsa_am1_3trj_pb0_gb2
| |
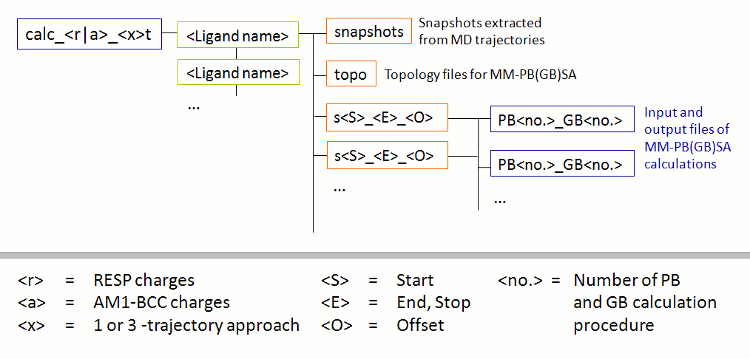
| 図 3:FEW によって作成されたMM-PB(GB)SAの計算のためのフォルダのサブ構造の模式図 |
MM-GBSA計算を開始するには calc_a_3t フォルダーにあるqsub_s51_100_1_pb0_gb2.sh スクリプトを実行する。 。
/home/user/tutorial/calc_a_3t/qsub_s51_100_1_pb0_gb2.sh計算の結果ファイルは、下記フォルダに表示される。
/home/user/tutorial/calc_a_3t/<ligandname>/s51_100_1/pb0_gb2
File: structs.txt
L51c
L51d
L51g
perl $FEW/miscellaneous/extract_WAMMenergies.pl structs.txt /home/user/tutorial/calc_a_3t pb0_gb2 51_100_1
最後の項は、"_"で区切られた first_PB_snapshot, last_PB_snapshot and offset_PB_snapshotsに対応している。 実行後、スクリプト pb0_gb2.txt が /home/user/tutorial/calc_a_3t フォルダーに作られ下記のように最終結果が要約される。| pb0_gb2.txt |
| Ligand ELE VDW NP_SOLV P_SOLV E_TOT L51c -192.36 -64.05 -4.80 189.55 -59.35 L51d -30.70 -62.01 -2.52 36.15 -48.68 L51g -103.96 -76.04 -4.92 113.28 -66.07 |
/home/user/tutorial/calc_a_3t/L51c/s51_100_1/pb0_gb2/L51c_statistics.out.snap
をみると、それぞれのスナプショットに対して計算された ΔGeffective値はca. -165.6 to +84.07 kcal/molに達することが分かる。 その結果、標準偏差は極めて高い(60.8キロカロリー/モル)。計算されたエネルギーの大きな変動に寄与する一つの要因は、内部エネルギーのノイズである。3-軌道によるアプローチでは、複合体、受容体、リガンドの内部エネルギーはキャンセルしないので、構造の間のわずかな差異が、計算された結合エネルギーに大きな影響を持ちうる。このノイズは、リガンドと受容体の遊離形態を無視し、結合エネルギーが複合体の構造のみに基づき算出された1軌道アプローチでは回避される。B: 1-軌道アプローチ
いわゆる1-軌道アプローチで、単に複合体のシミュレーションのみの構造が考慮され、結合対非結合の受容体およびリガンドと構造的な違いは考慮されていないかかわらず、このアプローチは、多くの場合適用され、良好な相対的な結合エネルギーの予測を得ことができることが示さている。このアプローチの大きな利点は、複合体だけのMDシミュレーションを実施するため、少ない計算で済むからである。それに加えて、内部エネルギー寄与のキャンセルによりノイズが少ないΔE値を得ることが出来る。| pb3_gb0.txt |
| Ligand ELE VDW NP_SOLV P_SOLV E_TOT L51c -19.05 -61.20 -6.20 61.16 -25.28 L51d -20.54 -64.81 -6.27 69.46 -22.17 L51g -21.64 -65.20 -6.28 66.50 -26.62 |
Copyright: Nadine Homeyer 2014