Tutorial A24
SECTION 4: 熱力学積分計算
FEWは、同じ受容体に結合するリガンドに対して熱力学的統合(TI)の手法により相対的結合自由エネルギーの決定を容易にする。プログラムは、必要な熱力学的統合(TI)変換シミュレーションの自動セットアップ機能を提供し、これらのシミュレーションの出力を用いて、ユーザーによる最小限の処置でリガンドの相対的な結合自由エネルギーが計算できる。
相対的結合自由エネルギーを決めるためのTIアプローチの基本原理が Tutorial A9中で説明されている。ここではもっぱらFEWによるTIの計算の設定を示した。FEWによるセットアップはTutorialA9で示されているものとは異なり、3種のシミュレーションではなく一つのシミュレーションがそれぞれのtransformationにたいして実行される。つまり、水中でのtransformationシミュレーションと溶媒和した複合体のなかでのtransformationシミュレーションである。
そのシミュレーションは単一のシミュレーションで電荷とVDW相互作用を変更することができる1ステップ、ソフトコア・アプローチに従ってFEWによって調製される (参照:[11]
およびAmber14マニュアルの355ページ)。
注意: TI の設定は事前に平衡化した構造を用いて行っているが、step 1Aに従ってパラメーターが用意され、例えば step 1Bにより溶媒和されたシステムが作られた複合体とリガンドの構造のTIの計算を開始することは原理的には可能である。平衡化されてない構造から、TIのシミュレーションを開始することを計画している場合FEWマニュアルを参照する 。
| |
| Figure 6: Transformation investigated by the TI approach. |
| TI_setup_L51c_d.in |
| @TIW ################################################################################ # Command file for TI simulation setup ################################################################################ # Location of input and output directories / file(s) # # lig_struct_path: Folder containing the ligand input file(s) # output_path: Basis directory in which all setup and analysis folders will # be generated. The directory must be identical with the # 'output_path' directory used for setup of the MD simulations. lig_struct_path /home/user/tutorial/structs output_path /home/user/tutorial ################################################################################ # Parameters required for TI simulation setup: # The following parameters have to be specified and need to be identical # in all subsequent runs for one system / TI-setup # # ti_simulation_setup: Request setup of TI simulation # charge_method: Charge method that shall be used, either "resp" or "am1" # lig_name_v0_struct: Name of start-ligand - Must be identical to the name of # the file in the "structs" folder used for generation of # parameter and library files with the common MD setup # functionality of FEW. # lig_name_v1_struct: Name of end-ligand - Must be identical to the name of # the file in the "structs" folder used for generation of # parameter and library files with the common MD setup # functionality of FEW. # lig_alias_v0: Alias that shall be used for the identification of the # start-ligand. The alias must consist of 3 characters. # lig_alias_v1: Alias that shall be used for the identification of the # end-ligand. The alias must consist of 3 characters. # softcore_mask_v0: Soft core atom mask for start-structure, specifying the # atoms of the start-structure (state V0) that shall be # regarded as soft core using the format # <lig_alias_v0>@<atom name list separated by comma> # softcore_mask_v1: Soft core atom mask for end-structure, specifying the # atoms of the end-structure (state V1) that shall be # regarded as soft core using the format # <lig_alias_v1>@<atom name list separated by comma> ti_simulation_setup 1 charge_method am1 lig_name_v0_struct L51c lig_name_v1_struct L51d lig_alias_v0 LFc lig_alias_v1 LFd softcore_mask_v0 LFc@C14,H1 softcore_mask_v1 LFd@N3 # ################################################################################ # 1) Parameters for preparation of coordinate and topology files of solvated # systems of start- and end-structures for TI simulations # # A) Generation of atom association list based on ligand mol2 files of # start and end structures # # prepare_match_list: Request creation of matching list prepare_match_list 1 # # B) Setup of coordinate and topology files # # It is required that RESTRT (coordinate) and topology files for the ligand and # complex of the start structure exist. These can be generated with the common # MD setup functionality of FEW. # # prepare_inpcrd_prmtop: Request setup of coordinate and topology files # lig_inpcrd_v0: Coordinate file (restart file) of ligand - start structure # com_inpcrd_v0: Coordinate file (restart file) of complex - start structure # lig_prmtop_v0: Topology of ligand - start structure # com_prmtop_v0: Topology of complex - start structure # match_list_file: Optional: File containing the atom association information # for the common part of start- and end-structures. Must only # be specified if step 1A was not successful and the list was # created manually. # chain_termini: Comma separated numbers of terminal residues of chains in # receptor structure. # create_sybyl_mol2: Request generation of mol2-files with sybyl atom types # for easy comparison of atom names of start- and end- # structures. Can facilitate checking and manual adjustment # of atom names in the end-structure, if automatic matching # is not successful. # additional_library: If an additional library file is required, e.g. for # non-standard residues present in the receptor structure, # this file must be specified here. # additional_frcmod: If additional parameters are needed, e.g. for describing # non-standard residues present in the receptor structure, # a parameter file should be provided here. # SSbond_file: File with disulfide bridge definitions # prepare_inpcrd_prmtop 1 lig_inpcrd_v0 /home/user/tutorial/MD_am1/L51c/lig/equi/md_nvt_red_06.restrt com_inpcrd_v0 /home/user/tutorial/MD_am1/L51c/com/equi/md_nvt_red_06.restrt lig_prmtop_v0 /home/user/tutorial/MD_am1/L51c/cryst/L51c_solv_lig.top com_prmtop_v0 /home/user/tutorial/MD_am1/L51c/cryst/L51c_solv_com.top match_list_file chain_termini 235 create_sybyl_mol2 1 additional_library /home/user/tutorial/input_info/CA.lib additional_frcmod SSbond_file /home/user/tutorial/input_info/disulfide_bridges.txt # 2) Setup scripts for TI MD # # General parameters # # no_shake: Set to "1", if no SHAKE shall be performed # ti_batch_path: Root path to be used in setup of batch files # ti_prod_template: Template script for TI production simulations no_shake 1 ti_batch_path /home/user/tutorial ti_prod_template /home/user/tutorial/input_info/MD_prod_noShake_TI.in # # A) Setup of scripts for equilibration # # ti_equil: Request generation of scripts for TI equilibration input # ti_equil_template: Template file for equilibration part of equilibration # phase of TI simulations. This equilibration part is # followed per default by a 1 ns free TI MD simulation # for complete equilibration of the system. # ti_equil_batch_template: Batch template file for equilibration phase of # TI simulations. # ti_equil_lambda: Values of lambda that shall be used in the calculation # in ascending order. Please specify only the decimal digits, # e.g. 1 for lambda 0.1, 05 for lambda 0.05. ti_equil 0 ti_equil_template /home/user/tutorial/input_info/equi_noShake_TI.in ti_equil_batch_template /home/user/tutorial/input_info/equi_TI.sge ti_equil_lambda 1,2,3,4,5,6,7,8,9 # # B) Setup scripts for production # # ATTENTION: This setup step can only be conducted if the equilibration # calculations have been completed. # # ti_production: Request generation of scripts for TI production input. # This setup step requires that the equilibration output is # present in the corresponding 'equi' folder. # ti_prod_lambda: Lambda steps for which the production shall be run; # separated by comma and in ascending order. Please specify # only the decimal digits, e.g. 1 for lambda 0.1. # total_ti_prod_time: Total production time requested (in ns) # ti_prod_batch_template: Batch template for TI production simulations # converge_check_script: Location of perl program to be used for convergence # checking after each production step. If the location # is not specified, it will be assumed that the program # can be found under the default location # at .../FEW/miscellaneous/convergenceCheck.pl # converge_check_method: Method that shall be used for convergence checking. # 1: Difference in standard error of dV/dL # 2: Precision of dV/dL according to student's # distribution # converge_error_limit: Error limit that shall be used as termination criterion # for the TI production simulations. # Defaults: 0.01 kcal/mol for method 1 # 0.2 kcal/mol for method 2 ti_production 0 ti_prod_lambda 1,2,3,4,5,6,7,8,9 total_ti_prod_time 1,1,1,1,1,1,1,1,1 ti_prod_batch_template /home/user/tutorial/input_info/prod_TI.sge converge_check_script converge_check_method 2 converge_error_limit 0.2 |
グローバルパラメータの定義
入力/出力ディレクトリ/ファイル(複数可)が定義されているコマンドファイルの最初の部分の主な用語は、section1で述べられたMDシミュレーションのセットアップのためのコマンドファイルの入力/出力指定と同じである。
TIのシミュレーション設定はリガンドのAM1-BCCチャージを使用して作成しなければならないことが明記されている。また、変換のために考慮しなければならないMOL2構造ファイルの名前、すなわちリガンドの名前、lig_name_v0_struct、 lig_name_v1_structが定義されている。対応するリガンドのエイリアス
lig_alias_v0と lig_alias_v1
がIDとして使用され、例えばリガンド残基名のファイル名に、表示される。
softcore_mask's
softcore_maskが 変更されなければならない原子、ソフトコア原子、すなわちリガンドL51cのリガンドL51dへの変更時に、デカップリングされ「見える」か「消える」かを変更する原子を定義する。
ステップ1:入力の座標とトポロジーファイルの準備
TIミュレーションのための座標とトポロジーファイルのセットアップをするために、ti_simulation_setupと and prepare_inpcrd_prmtopのフラグを1に設定する。
A: 実際の入力ファイルを用意する前に、両方のリガンドで構造的に等価である原子はペアとしてリストされている原子の関連(association)リストが作成される。のちにリガンドのすべての共通原子が座標ファイルで同じ順序でリストする必要があるため等価の原子の識別が必要である。したがって、正しい原子関連(association)リストは、シミュレーションの成功のセットアップのために不可欠である。通常、このステップは、ユーザーの介入なしに行われている。
B:
原子関連(association)リストが作成されると、実際の座標とトポロジーファイルが生成される。変換の終了状態のファイル(このケースでL51dである)が、開始状態の座標とトポロジーファイルに基づいて作成される。このように、溶媒和リガンドL51c(lig_inpcrd_v0)および溶媒和コンプレックスの(com_inpcrd_v0)の座標ファイルが必要である。座標ファイルは、このチュートリアルのsection
1 で調製されたMDシミュレーションの最終平衡段階から得られる。シミュレーションに使用される対応するトポロジーファイルはシミュレーションディレクトリのcrystフォルダーに存在し、コマンドファイルのlig_prmtop_v0と com_prmtop_v0で設定されている。
タンパク質Xa因子は、モノマーではなく、二つの鎖で構成されているので、最初のチェーン末端はchain_terminiで定義されている。受容体の他の特定の機能は、section 1で示したように、additional_library、 additional_frcmod、と SSbond_file設定を介して処理することができる。(section 1を参照)
FEW実行:
basic input/output path、すなわち/home/user/tutorialをチュートリアルファイルが保存されているローカルシステムにパスに変更してあれば、FEWは、以下のコマンドで実行出来る:
perl $FEW/FEW.pl TI /home/user/tutorial/cfiles/TI_setup_L51c_d.in
上で指定されたコマンド・ファイルを ti_simulation_setup と prepare_inpcrd_prmtop を 1と設定してFEW実行すると 新しいフォルダTI_am1が/home/user/tutorialディレクトリー内に作成されている。このフォルダTI_am1 には、コマンド・ファイルでlig_name_v0_struct と lig_name_v1_struct のように特定のリガンドの名前にちなんで名付けられたサブフォルダが含まれる。 上記のコマンド・ファイルで指定された名前の場合、フォルダーはL51c_L51dという名前が付けられる。 このサブフォルダに、この特定の変換に関連付けられているすべてのデータが(図7)に保存される。現在、セットアップ フォルダには、セットアップ時に生成された座標とトポロジーファイルが含まれている。
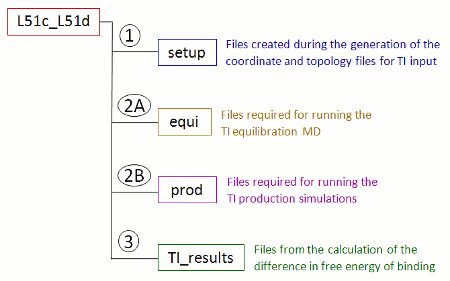 |
| 図 7: FEW とTIの計算のセットアップ時に作成した変換特定のフォルダのサブ構造の模式図。 |
ステップ2:TIのMDシミュレーションのセットアップ
A:
平衡
TIのMDシミュレーションの平衡化段階のためのファイルはti_equilを
1を設定し準備できる。 シミュレーションは、水素原子を含む拘束結合のためのシェイクアルゴリズムを使用せずに調製する(no_shake=1)。 output_pathと定義されたパスは、同時に準備されたバッチスクリプトのための入力/出力パスとして使用されている(ti_batch_path)。
シミュレーションの平衡化段階に対する入力ファイルは、ti_equil_template と ti_prod_templテンプレートを用いて9 &lambda、値 0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、 0.9、のため作られる。この段階で、 ti_equi_templの設定に従って純粋な平衡ステップが、続いて ti_prod_templを用いて1 ns固定のシミュレーションが平衡を完全にするために行われている。バッチテンプレート ti_equi_batch_templateは使用する前に、Fix variablesステートメントと>re-queuing題されたセクションの前のトップセクションを変更しローカル・コンピューティングに適合させる必要がある。
qsub /home/user/tutorial/TI_am1/L51c_L51d/equi/com/run.pbs
異なるλ値に対する平衡化過程は、図8に示されるスキームに従って順次処理される。
 |
| 図 8: TI シミュレーションの平衡化と生産段階のワークフローの概略図。 |
注意:
平衡化シミュレーションが生産段階(production phase)で考慮されなければならないすべてのλの値について完了した場合は、下記の生産TI MDシミュレーションのセットアップを起動することができる。平衡シミュレーションの実行には時間がかかるので、シミュレーション中に記録されたエネルギーの出力ファイルは、FEWチュートリアルフォルダに保存される。保存されたMDの出力ファイルを使用する場合は、link_TIequi_output.shファイルをbasic input/output folderにコピーする。
cp $FEW/examples/tutorial/link_TIequi_output.sh /home/user/tutorial
そしてパスをFEWがローカルシステムにインストールされているパスの3行目にある /home/src/FEW に変更する。次に、スクリプトを実行する。
/home/user/tutorial/link_TIequi_output.sh
そして、シェルから出て、ソフトリンクが/home/user/tutorial/TI_am1/L51c_L51d/equi にあるcomとligフォルダーに作成されていることを確認する。
B:
Production
変換シミュレーションのproduction部分の入力ファイルの作成は ti_productionを1
に設定することによって実行する。シミュレーションのセットアップは、平衡計算が実行されたすべての9λステップで要求される。また、equilibrationsが実行されているそれらのλの一部のproduction setupのセットアップをすることも可能である。しかし、Δλの大きさ、つまりλのステップサイズは、すべてのλ対して等しくなければいけない。したがって、λ 0.2、0.4、0.6、および0.8値、のためのシミュレーションを実行することも可能である。TI のproductionシミュレーションは平衡からの最終座標ファイルに基づいて開始される。(student分布で95%の信頼水準での)0.2キロカロリー/モルよりも良好な精度が、シミュレーションの終了基準として使用される。この基準に達していない場合は、すべてλの値でシミュレーションはtotal_ti_prod_tim=1 ns後に終了する。
バッチ・テンプレート・ファイル ti_prod_batch_template
は 上記のti_equil_batch_templateファイルと同じように適応させると、TIのproductionジョブ実行のバッチファイルのセットアップ用テンプレートファイルのとして機能する。
FEWの実行:
コンピューター特異的な環境を整えた後、
コマンドファイルにあるti_productionをスイッチオンすることによりFEWのTI production simulationsセットアップは以下のようにして実行する。
perl $FEW/FEW.pl TI /home/user/tutorial/cfiles/TI_setup_L51c_d.in
セットアップは、逆累積平均化手順に従って平衡の検査を行い[12,13]、システムが平衡化され、TIのproductionに使用することができることを確認する。チェックルーチンが完了した後、平衡チェックが成功した旨の表示がある。本ケースの場合、平衡化チェックがすべてのλに対してリガンドと複合体システムの両方でパスしている。全体的な平衡チェックではパスするが、平衡チェックが個々λでは失敗することは起こりうる。この場合、それぞれのequilibrationsは後で結合自由エネルギー予測の精度を下げる事になる平衡化中の問題を完全に取り除くために分析されるべきである。DV/dλ値 対 シミュレーション時間のプロットは、このような問題を識別するための重要である。図9は、出力ファイルから抽出したdV/dλ値とそのcummulative平均(赤線)のシミュレーション時間に対するプロットを示している。DV /dλの値が大幅に変動するが、cummulative平均値が比較的一定であることは明らかである。これは、システムが平衡化された状態で既に平均値であり、そして別のλ値で同じ精度に達するために必要とされることはただ長いサンプリング値であることを示している。production実行の精度レベルの制御は実装されている収束チェックを介して確保されているので、平衡の最終的な構造は、このような場合には、productionのための入力構造として使用することができる。
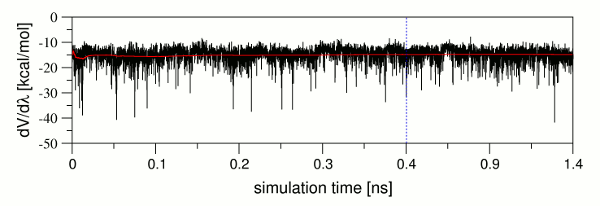 |
| 図
9: DV/dλ(黒)とcummulative平均(赤)vs シミュレーション時間のプロット シミュレーションの純粋な平衡部分が終了し、平衡化フェーズの1ナノ秒のproduction開始のポイントは青の破線で示されている。 |
TIのproductionシミュレーションのファイルはprodという名前のフォルダ内に保存されている。このフォルダには、上記のequiフォルダと同様に、溶液中のリガンドおよび溶媒和複合体のシミュレーションのため、二つのサブフォルダ、ligとcom、が含まれている。 productionシミュレーションはバッチスクリプトを実行することによって開始する:
/home/user/tutorial/TI_am1/L51c_L51d/qsub_prod.sh
注Note: 9個のλ値について2つのシステムのためにTIのシミュレーションを行うことは計算的に煩雑であるので、エネルギー値を含むシミュレーションの出力ファイルは、FEWで与えられる。これらのファイルを使用する場合は、スクリプト copy_TIprod_output.sh をtutorial folder にコピーし、
cp $FEW/examples/tutorial/copy_TIprod_output.sh /home/user/tutorial
そしてパス /home/src/FEW を change the path FEWがインストールされている/home/src/FEW ディレクトリに変更し、スクリプトを実行する。
/home/user/tutorial/copy_TIprod_output.sh
スクリプトの実行を終えた後、 シェルを出てTI productionの出力ファイルが/home/user/tutorial/TI_am1/L51c_L51d/prodにある lig と com フォルダーにコピーされていることを確認する。
ステップ 3 : 相対的結合自由エネルギーの計算
L51cとL51dの結合自由エネルギーの差、ΔΔGbindingは、TIの生産シミュレーションからの出力に基づいて自動化された方法でFEWを用いて計算することができる。コマンドファイルTI_setup3_L51c_d.in は、 この作業に必要なパラメータを含んでいる。| TI_setup3_L51c_d.in |
| @TIW ################################################################################ # Command file for calculating the difference in binding free energy # between start and end ligand based on TI simulations. ################################################################################ # Location of input and output directories / file(s) # # lig_struct_path: Folder containing the ligand input file(s) # output_path: Basis directory in which all setup and analysis folders will # be generated. The directory must be identical with the # 'output_path' directory used for setup of the MD simulations. lig_struct_path /home/user/tutorial/structs output_path /home/user/tutorial ################################################################################ # Parameters required for TI analysis: # # ti_ddG: Request calculation of difference in binding free energy. # charge_method: Charge method that shall be used, either "resp" or "am1" # lig_name_v0_struct: Name of start-ligand - Must be identical to the name of # the file in the "structs" folder used for generation of # parameter and library files with the common MD setup # functionality of FEW. # lig_name_v1_struct: Name of end-ligand - Must be identical to the name of # the file in the "structs" folder used for generation of # parameter and library files with the common MD setup # functionality of FEW. # lig_alias_v0: Alias that shall be used for the identification of the # start-ligand. The alias must consist of 3 characters. # lig_alias_v1: Alias that shall be used for the identification of the # end-ligand. The alias must consist of 3 characters. # dVdL_calc_source: Specification of files considered in computation of ddG: # 0: All files available for ligand and complex are considered # X-Y: Where X is the number of the first and Y is the number # of the last production file that shall be taken into account. # If Y=0 all files from X on will be considered. # ddG_calc_method: Method to be used for the calculation of dG: # 1: Conventional numerical integration with interpolation # to lambda=0 and lambda=1. # 2: Numerical integration without interpolation # ti_ddG 1 charge_method am1 lig_name_v0_struct L51c lig_name_v1_struct L51d lig_alias_v0 LFc lig_alias_v1 LFd dVdL_calc_source 0 ddG_calc_method 1 |
FEWがTI_setup3_L51c_d.inコマンドファイルにより実行された後、TI_results と呼ばれる新しいフォルダが transformation specificのディレクトリ(図7)に出来ている。このフォルダには、ΔΔGbindingの計算で考慮されるテキストファイルであるすべてのdV/dλ値リストと、ログファイルと最終の結果が保存されるTI_dG.outが含まれる。
L51cとL51d間の結合の自由エネルギーの計算による違いは2.00キロカロリー/モルである。1ナノ秒という短い時間のシミュレーションであり、これは非常に大まかな結合自由エネルギーの見積もりで、 dV/dλはすべてのλに対して望むべきエラーリミットに達していないからである。特に複雑な状態ではthe λの 0.8 と 0.9に関してはエラーは依然として高い。またここで用いられたtransformationは非常に小さく、そのためproductionシミュレーションは比較的早く収束するが、心に留めておくべきことは他の変換では遥かに長いシミュレーションが必要かもしれないことである。
SECTION 1: Setup of MD simulations
SECTION 2: MM-PB(GB)SA calculations
Copyright: Nadine Homeyer 2013